






SIRT6调节的巨噬细胞胞葬表观遗传控制糖尿病牙周炎的炎症消退

ž 糖尿病是一种以胰岛素抵抗、高血糖和炎症紊乱为特征的疾病,会导致严重的牙周炎。与慢性牙周炎(CP)相比,糖尿病性牙周炎(DP)对牙周组织的破坏更为严重。DP的高糖环境会导致宿主免疫炎症反应失调,从而引发持续炎症,加速牙周破坏。糖尿病患者的慢性不愈合或愈合缓慢的伤口通常与长期炎症和中性粒细胞持续性有关。此外,随着胞葬的生物学行为,巨噬细胞分泌大量抗炎分子,表明这一过程是促进炎症消退和组织修复所必需的。新出现的证据强调了胞葬受损在多种炎症性疾病中的作用,而胞葬在DP中的生理功能尚未完全确定。SIRT6作为一种NAD依赖性组蛋白去乙酰化酶,在糖代谢、炎症稳态和长寿中具有转录调节功能。髓细胞特异性SIRT6缺乏通过调节巨噬细胞表型,延迟高脂饮食小鼠的伤口愈合并引起胰岛素抵抗。此外,对糖尿病患者的研究表明,SIRT6在许多人体组织中表达不足,这表明SIRT6对糖尿病相关疾病具有保护作用。因此,探索SIRT6调控免疫稳态的潜在机制,可能对DP患者实现良好的牙周组织保存具有重要的临床意义。该研究发表于《Theranostics》,IF:11.6。
技术路线:

主要研究结果:
1. 人DP中中性粒细胞和巨噬细胞的特征分布
为了探索糖尿病相关炎症性疾病中细胞群组成和调节细胞通路的差异,作者在单细胞测序数据集GSE165816中分析了来自健康非糖尿病和糖尿病足溃疡非愈合者的足部皮肤样本。GO富集分析结果显示,炎症和细胞凋亡相关通路被激活(图1A, B)。在鉴定的炎症细胞中,作者重点关注巨噬细胞浸润增加,并通过GO分析和统一流形逼近与投影(UMAP)分析进一步鉴定巨噬细胞簇的细胞群。值得注意的是,UMAP分析显示巨噬细胞异常极化(图1C)。GO富集分析显示,与非糖尿病对照的巨噬细胞相比,糖尿病巨噬细胞中与免疫应答、吞噬、迁移能力和葡萄糖代谢相关的通路出现异常(图1D, E)。这些结果表明,糖尿病创面愈合过程中异常炎症反应可能是由于细胞凋亡相关通路大量激活,巨噬细胞吞噬能力异常所致。
为了进一步探讨DP持续升高的炎症是否与巨噬细胞吞噬能力异常有关,作者收集了伴有或不伴有糖尿病的牙周炎患者的牙龈样本(图1F)。在牙周炎中,巨噬细胞引起的中性粒细胞胞葬被认为是抗炎和促进消退的事件。糖尿病的慢性炎症通常很严重且很难治愈。因此,作者首先检测了牙龈中的中性粒细胞,结果显示DP组的中性粒细胞浸润(CD15)细胞数量高于CP组(图1G, L)。使用牙龈组织连续切片进行TUNEL染色,结果显示DP组的牙龈组织中有大量凋亡细胞,而CP组几乎未见凋亡细胞(图1H, M)。通过对相同位点的连续切片的比较观察,作者发现大多数凋亡细胞为CD15中性粒细胞(图1G, H)。随后,作者利用MPO和H3CIT标记物中性粒细胞胞外诱捕网(NETs)发现,DP中中性粒细胞的过度积累和延迟清除导致大量NETs的形成(图1I, N),从而加重了糖尿病患者的炎症并延迟了炎症的消退。为了进一步探讨未及时清除的凋亡中性粒细胞是否由巨噬细胞浸润异常引起,作者对巨噬细胞极化进行免疫荧光染色。DP组牙龈中巨噬细胞(CD68)的浸润量远高于CP组(图1O)。DP组CD68 CD86阳性M1巨噬细胞浸润量高于CP组(图1J, P),CD68 CD206阳性M2极化巨噬细胞浸润量DP组与CP组无差异(图1K, Q),但DP组M2/M1比值明显低于CP组(图1R)。综上所述,凋亡中性粒细胞和NETs的过度积累和延迟清除,伴随着巨噬细胞浸润增加和巨噬细胞极化破坏,无疑加重了DP炎症。
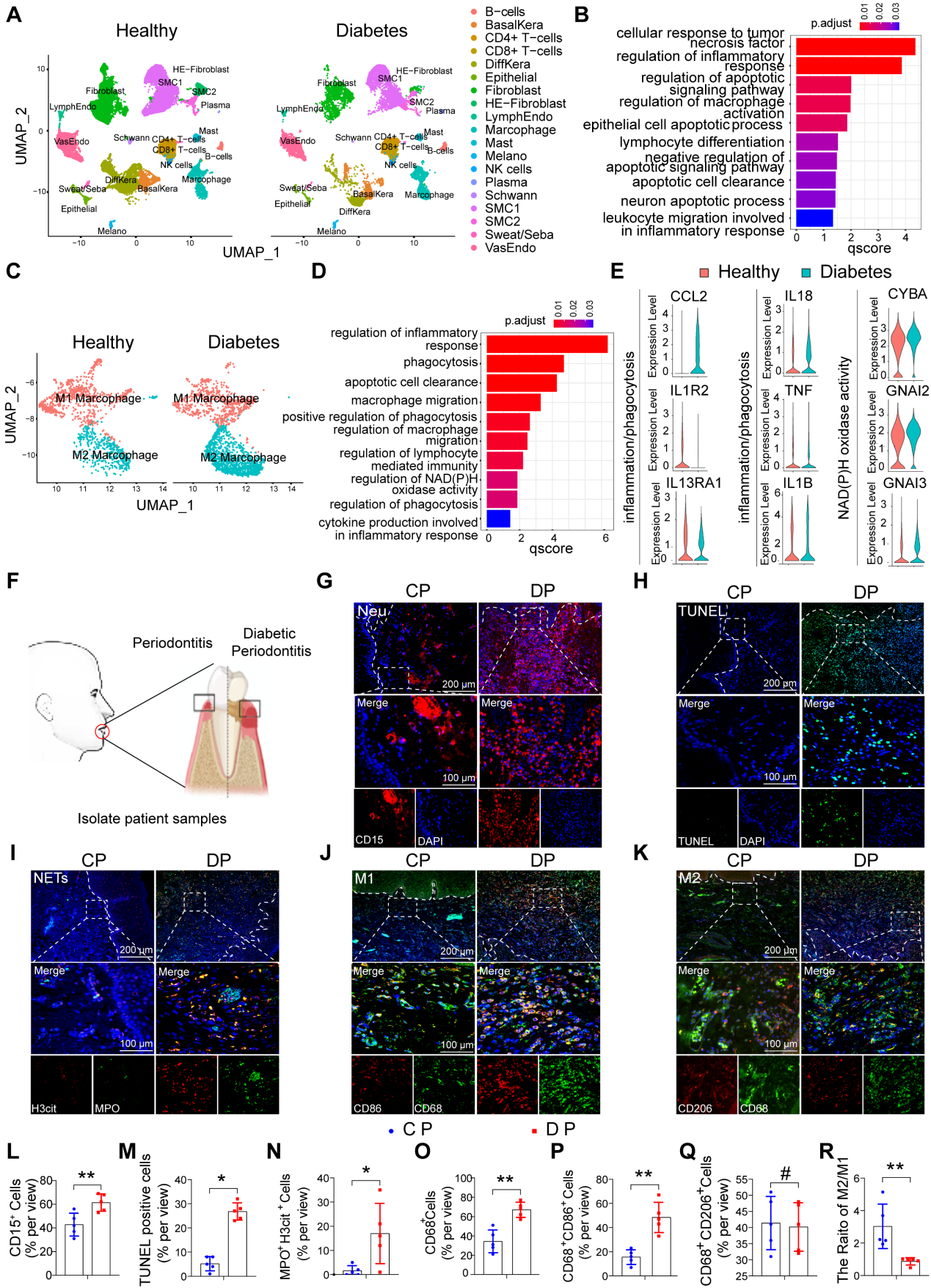
图1 人DP中中性粒细胞和巨噬细胞的特征分布
2. 功能失调的中性粒细胞和巨噬细胞加重了小鼠DP的炎症损伤并损害炎症消退
为了探讨糖尿病对牙周炎进展和炎症消退的影响,作者使用了两种经典的结扎性牙周炎(LIP)小鼠模型,即牙周炎模型和牙周炎消退模型。牙周炎模型结扎过程长达14天,牙周炎消退模型小鼠结扎7天,然后拆除结扎物,再模拟7天的炎症消退过程(图2A)。在牙周炎和牙周炎消退模型中,糖尿病小鼠从CEJ到ABC的距离都明显增加(图2B, I)。在牙周炎和牙周炎消退模型中,与CP组相比,糖尿病小鼠表现出更高的破骨细胞活性,TRAP阳性破骨细胞表面出现更高(图2C, J)。此外,在DP牙周组织中可见大量中性粒细胞浸润、MPO和H3cit NETs(图2D, K, E, L)。TUNEL染色也显示DP牙周组织中有大量凋亡细胞,而CP组未见明显凋亡细胞(图2F, M)。DP中F4/80巨噬细胞浸润数量大于CP组(图2N)。具体而言,DP组F4/80 CD86 M1巨噬细胞(图2G, O)和F4/80 CD206 M2巨噬细胞(图2H, P)数量明显高于CP组。然而,在LIP模型的分辨率上,DP组的M2/M1巨噬细胞比例也明显低于CP组(图2Q)。总的来说,凋亡中性粒细胞和NETosis在牙周组织中的积累加速了牙周炎的进展,而凋亡中性粒细胞和NETs的延迟清除阻碍了小鼠DP炎症的消退。

图2 功能失调的中性粒细胞和巨噬细胞加重了小鼠DP的炎症损伤并损害炎症消退
3. SIRT6显著调节高糖条件下巨噬细胞的胞葬作用
鉴于上述结果,作者进一步解读HG是否抑制巨噬细胞对凋亡中性粒细胞的胞葬作用,从而间接增加其残基。HG刺激后巨噬细胞吞噬凋亡中性粒细胞的能力下降(图3A)。鉴于DM中巨噬细胞簇的GO富集分析显示NAD代谢异常(图1D, E),作者进一步检查发现HG刺激可导致NAD/NADH代谢异常(图3B)。Sirtuins(SIRTs)是NAD依赖性组蛋白去乙酰化酶家族,在葡萄糖稳态、炎症、基因组稳定性和DNA修复中发挥作用。因此,由于SIRT6在HG条件下明显下调,在48 h和72 h时最为明显,作者对其进行了鉴定和重点研究(图3C)。为了进一步研究SIRT6在DP巨噬细胞中的可能功能,作者将巨噬细胞标志物CD68和SIRT6共染色,发现DP巨噬细胞中SIRT6的表达与CP相比显著降低(图3D)。SIRT6抑制剂显著降低了巨噬细胞进行胞葬的能力,而SIRT6过表达提高了巨噬细胞吞噬凋亡中性粒细胞的能力(图3E, F)。综上所述,高糖刺激导致巨噬细胞SIRT6的低表达,从而损害了胞葬作用。胞葬通过减少死细胞的DAMP释放和维持生物稳态来抑制炎症。然而,当糖尿病患者巨噬细胞的胞葬作用受损时,通过损伤相关分子识别模型,包括NETosis和坏死性凋亡在内的促炎溶解细胞死亡,导致巨噬细胞浸润增加,打破了M1和M2分化的平衡。

图3 SIRT6显著调节高糖条件下巨噬细胞的胞葬
4. 骨髓特异性SIRT6敲除会加重牙周炎并损害炎症消退
为了探索SIRT6介导的巨噬细胞胞葬在牙周炎中的作用,作者将LysM-Cre小鼠与SIRT6flox/flox小鼠杂交产生mS6KO小鼠。与同窝野生型小鼠骨髓巨噬细胞(BMMs)相比,mS6KO骨髓巨噬细胞吞噬凋亡中性粒细胞的能力下降(图4A)。采用2月龄mS6KO小鼠及其WT窝代小鼠建立LIP模型和LIP分辨率模型。与WT小鼠相比,CEJ到ABC的距离(图4B, I)和捕获阳性破骨细胞在mS6KO小鼠中显著增加(图4C, J)。为了研究巨噬细胞胞葬受损是否参与了mS6KO小鼠的持续性骨质流失,通过TUNEL染色评估mS6KO小鼠和WT小鼠牙周组织中凋亡细胞的数量(图4D),结果显示,与同窝野生型小鼠相比,mS6KO小鼠牙周组织中Ly6g细胞和凋亡细胞的数量显著增加(图4E, L)。SIRT6敲除促进了mS6KO小鼠牙周组织中MPO H3cit的NETosis(图4F, M)。此外,F4/80 CD86 M1巨噬细胞(图4G, O)和F4/80 CD206 M2巨噬细胞(图4H,P)在mS6KO小鼠牙周组织中显著升高(图4N),但抗炎F4/80 CD206 M2巨噬细胞在mS6KO小鼠牙周组织中的比例低于WT小鼠(图4Q)。综上所述,这些发现表明SIRT6介导的巨噬细胞胞葬与牙周破坏和炎症消退有关。

图4 髓细胞特异性SIRT6基因敲除会加重牙周炎并损害炎症消退
5. SIRT6通过H3K56ac抑制miR-216a-5p-216b-5p-217簇的转录
为了深入了解SIRT6调控巨噬细胞胞葬的详细机制,作者进一步评估了SIRT6对巨噬细胞中microRNA表达的影响。从SIRT6抑制后的巨噬细胞中分离RNA进行miRNA PCR芯片分析,结果显示miR-216a-5p-216b-5p-217簇明显增加,SIRT6表达下调(图5A, B, C)。最近的研究表明,miR-216/217在动脉粥样硬化和糖尿病等年龄相关疾病中起关键作用。与这些结果一致,作者的数据进一步显示SIRT6抑制和HG条件下miR-216a-5p-216b-5p-217簇表达增加(图5D, F),SIRT6过表达下表达降低(图5E)。为了进一步阐明SIRT6如何抑制miR-216a-5p-216b-5p-217簇的表达,作者通过ChIP-qPCR检测了巨噬细胞中SIRT6宿主基因MIR217HG启动子的组蛋白修饰。SIRT6通过催化位点3和9乙酰化的组蛋白H3赖氨酸残基(H3K9ac和H3K56ac)32的去乙酰化而发挥转录共抑制作用。有趣的是,作者发现HG处理仅显著上调H3K56ac的表达(图5H)。使用覆盖miR-216a-5p-216b-5p-217启动子的7对引物进行检测,结果显示HG处理增加了miR-216a-5p-216b-5p-217集群启动子的H3K56ac水平(图5I)。作者还进一步证实了SIRT6抑制和HG条件下pri-miR-217簇表达增加(图5J, L),SIRT6过表达后pri-miR-217簇表达下调(图5K)。值得注意的是,来自mS6KO小鼠的BMMs表现出增加的pri-miR-217和miR-216a-5p-216b-5p-217簇表达(图5G, M)。

图5 SIRT6通过H3K56ac抑制miR-216a-5p-216b-5p-217簇的转录
6. SIRT6通过“非规范”微处理器复合物抑制miR-216a-5p-216b-5p-217簇成熟
典型microRNA的成熟最初是初级microRNA(pri-miR)转录物的转录,该转录物由核糖核酸酶III、Drosha(核糖核酸酶)和DGCR8组成的微处理器复合物鉴定和切割。有报道称,葡萄糖代谢影响Drosha蛋白的表达,葡萄糖剥夺促进Drosha表达,HG刺激抑制Drosha表达。同样,作者的结果显示HG刺激可以抑制巨噬细胞中Drosha蛋白的表达(图6A)。尽管Drosha降低,但作者发现高葡萄糖导致miR216a-5p-216b-5p-217簇的高表达(图5F)。为了进一步探索HG条件下miR216a-5p-216b-5p-217簇成熟的机制,作者设计了一种特异性的生物素标记的pri-miR-217探针,在巨噬细胞中进行RNA沉淀实验,银染色显示与pri-miR-217结合的多条蛋白带富集(图6B)。同时,蛋白质谱分析显示hnRNPA2B1在识别蛋白列表中排名靠前,未观察到Drosha和DGCR8。RIP实验显示,与IgG相比,hnRNPA2B1抗体降低了更多的pri-miR-217(图6C)。据报道,hnRNPA2B1与microRNA微处理器复合物蛋白DGCR8相互作用,通过结合m6A标记初级miRNA转录物来切割初级miRNA。内源性hnRNPA2B1蛋白与巨噬细胞中DGCR8和Drosha的共免疫沉淀(Co-IP)表明它们之间存在直接的物理相互作用,hnRNPA2B1与DGCR8结合,而未观察到Drosha(图6F)。作者进一步确定DGCR8可以与Drosha结合,这意味着hnRNPA2B1与DGCR8结合以招募Drosha形成微处理器复合物,“非规范”微处理器复合物识别并切割pri-miR-217(图6E)。Co-IP还显示SIRT6抑制促进了“非规范”微处理器复合物的形成,SIRT6过表达减少了微处理器复合物(图6G)。此外,作者进一步发现,高糖增加了与pri-miR-217结合的hnRNPA2B1(图6D)。Chip的结果也显示HG处理增加了hnRNPA2B1启动子的H3K56ac(图6H)。为了确定HG是否以hnRNPA2B1依赖性方式影响pri-miR-217的加工,作者对巨噬细胞进行了hnRNPA2B1敲低,发现成熟miRNA的水平下降,而pri-miR-217的水平增加(图6I, J)。FISH染色显示HG刺激导致细胞核中pri-miR-217的减少,敲低hnRNPA2B1后细胞核中pri-miR-217的积累(图6K)。这些数据表明SIRT6不仅可以促进pri-miR-217的转录,还可以通过促进HG条件下“非规范”微处理器复合物的形成来促进miR216a-5p-216b-5p-217簇的成熟。
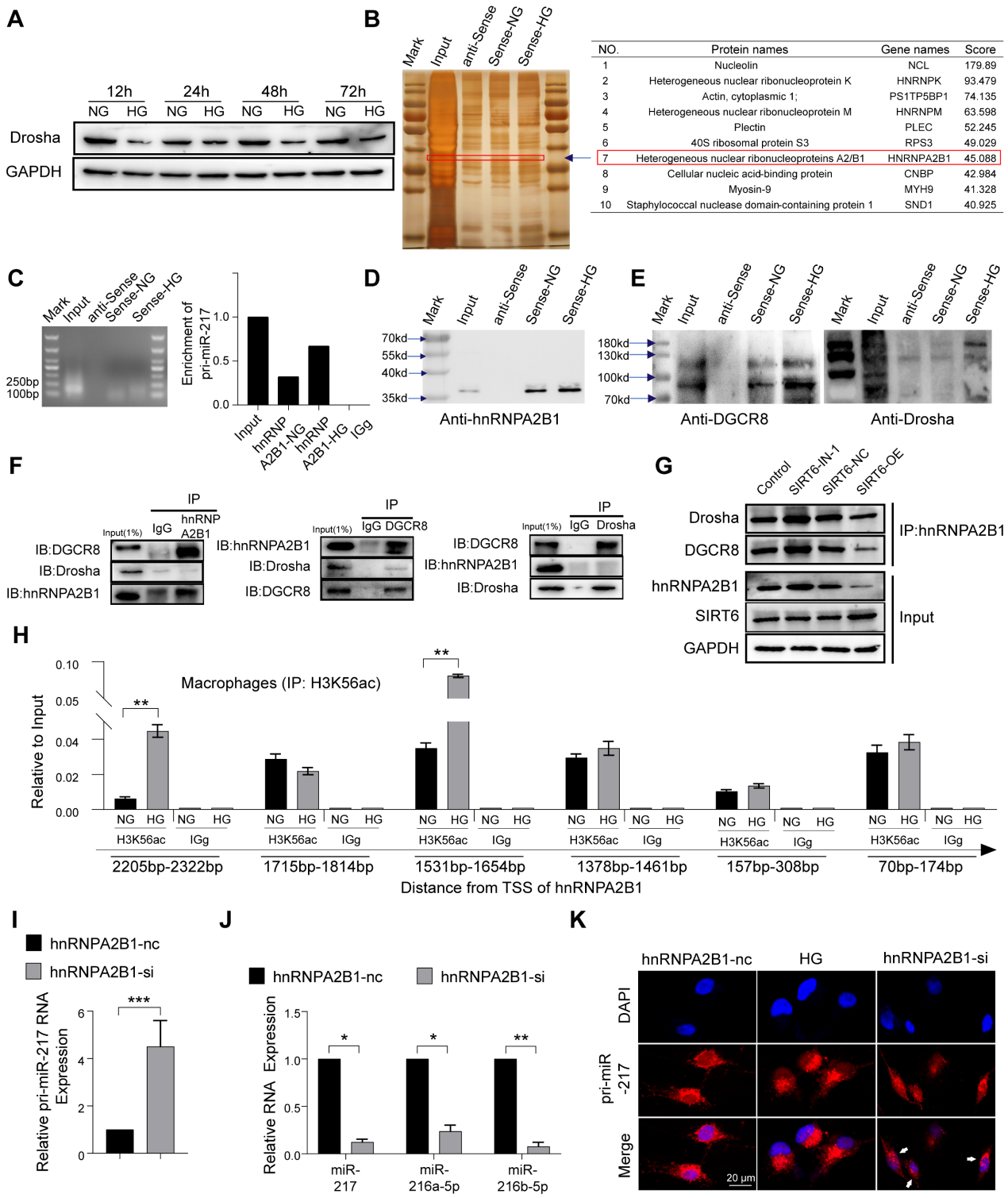
图6 SIRT6通过“非规范”微处理器复合物抑制miR-216a-5p-216b-5p-217簇成熟
7. miR-216a-5p-216b-5p-217簇通过靶向DEL-1和CD36负调节巨噬细胞的胞葬作用
DEL-1和CD36已被确定为巨噬细胞胞葬和清除炎症的关键调节分子。生物信息学算法(miRwalk)的结果表明,miR-216a-5p-216b-5p-217簇与DEL-1和CD36 mRNA的3'UTR结合(图7A)。随后,双荧光素酶报告基因实验显示,miR216a-5p-216b-5p-217簇的共转染降低了WT-DEL-1-3'UTR和WT-CD36-3'UTR的荧光素酶活性,而Mut-DEL-1-3'UTR和Mut-CD36 -3'UTR的荧光素酶活性不受影响(图7B)。此外,蛋白质印迹分析显示,在miR216a-5p-216b-5p-217 mimic处理的巨噬细胞中,DEL-1和CD36的表达降低,而在miR-216b-5p-217 inhibitor处理的巨噬细胞中,DEL-1和CD36的表达升高(图7C)。免疫荧光也证实了DEL-1和CD36在糖尿病牙周炎中的低表达(图7D)。miR216a-5p-216b-5p-217簇的过表达降低了巨噬细胞的吞噬能力(图7E)。在这三种microRNA中,miR-217对巨噬细胞吞噬作用的影响最大。在HG环境下敲低miR-217可以恢复巨噬细胞的胞葬,与重组DEL-1蛋白的作用类似(图7F)。总体而言,作者的研究结果支持SIRT6通过促进pri-miR-217转录和pri-miR“非规范”微处理器复合物的形成负调控miR216a-5p/216b-5p-217簇表达的假设。该microRNA簇通过靶向关键的胞葬分子DEL-1和CD36抑制巨噬细胞的胞葬,导致巨噬细胞介导的炎症消退功能失调。
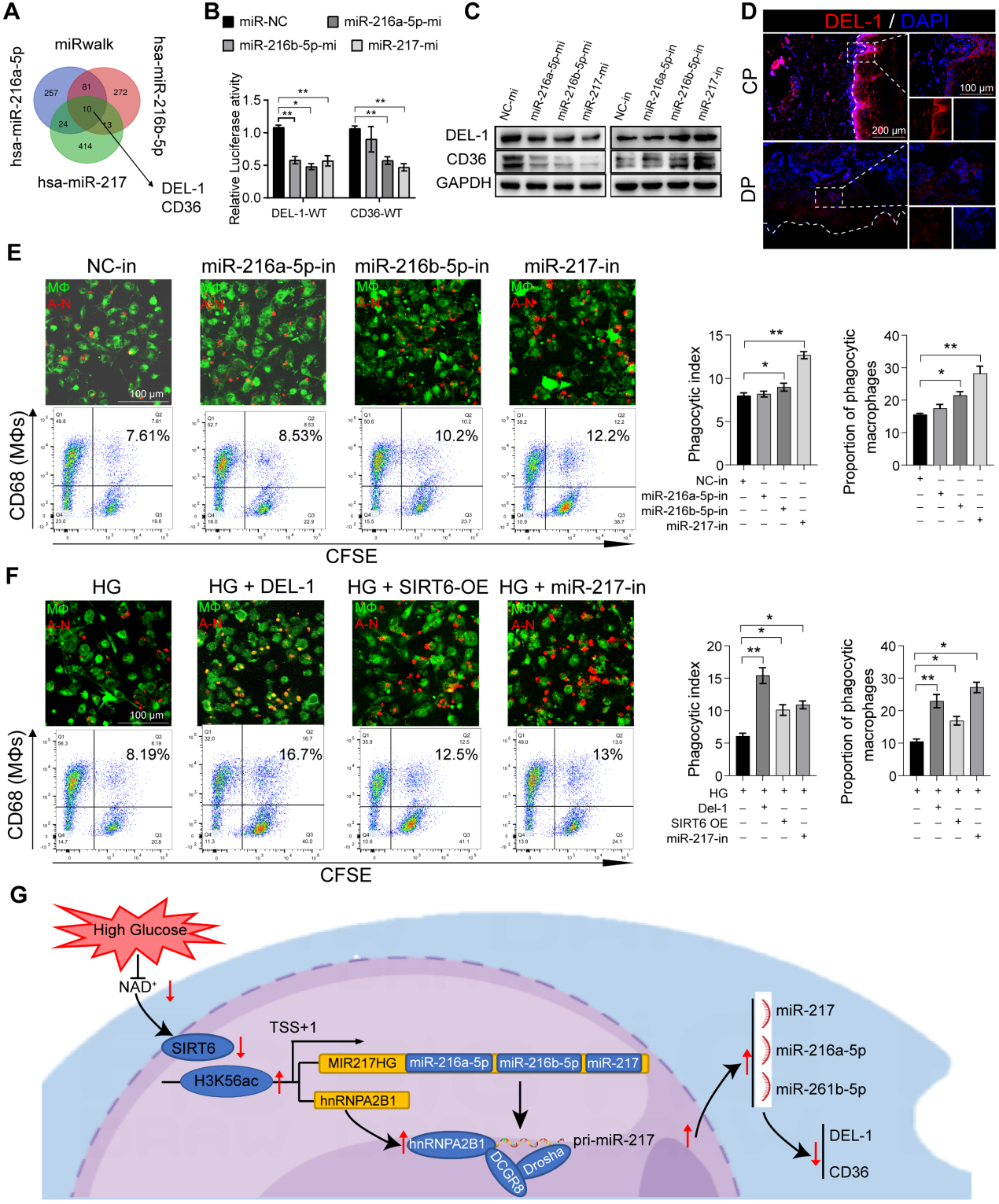
图7 miR-216a-5p-216b-5p-217簇通过靶向DEL-1和CD36负性调节巨噬细胞的胞葬
8. antagomir-217的局部给药促进小鼠DP炎症的消退
根据上述功能获得和功能丧失研究中的吞噬指数观察,miR-217表现出更好的表型,并被选择用于体内干预治疗。作者在结扎丝后第3、6、9和12天立即将miR-217特异性拮抗剂或scramble(NC)-miR注射到糖尿病小鼠的牙周组织中(图8A)。在第7天解除结扎后,antagomir-217注射明显改善了糖尿病小鼠牙周炎症和骨质丢失(图8B, C, G, H)。注射antagomir-217后,糖尿病小鼠牙周组织中DEL-1的表达明显增加(图8D, I)。注射antagomir-217后,糖尿病小鼠牙周组织中Ly6g中性粒细胞的数量明显减少(图8E, J)。注射antagomir-217后,MPO H3cit NET(图8L)和凋亡细胞的形成明显减少(图8F, K)。antagomir-217治疗组M2型抗炎巨噬细胞(图8O)比例较高,而对照组大部分巨噬细胞为M1型巨噬细胞(图8M, N, P)。总而言之,这些结果表明,通过沉默miR-217抑制SIRT6 / miR216a-5p-216b-5p-217簇/ DEL-1和CD36调节轴可促进巨噬细胞的胞葬以清除凋亡细胞,这意味着该调节轴具有作为糖尿病炎症解决靶点的潜力。
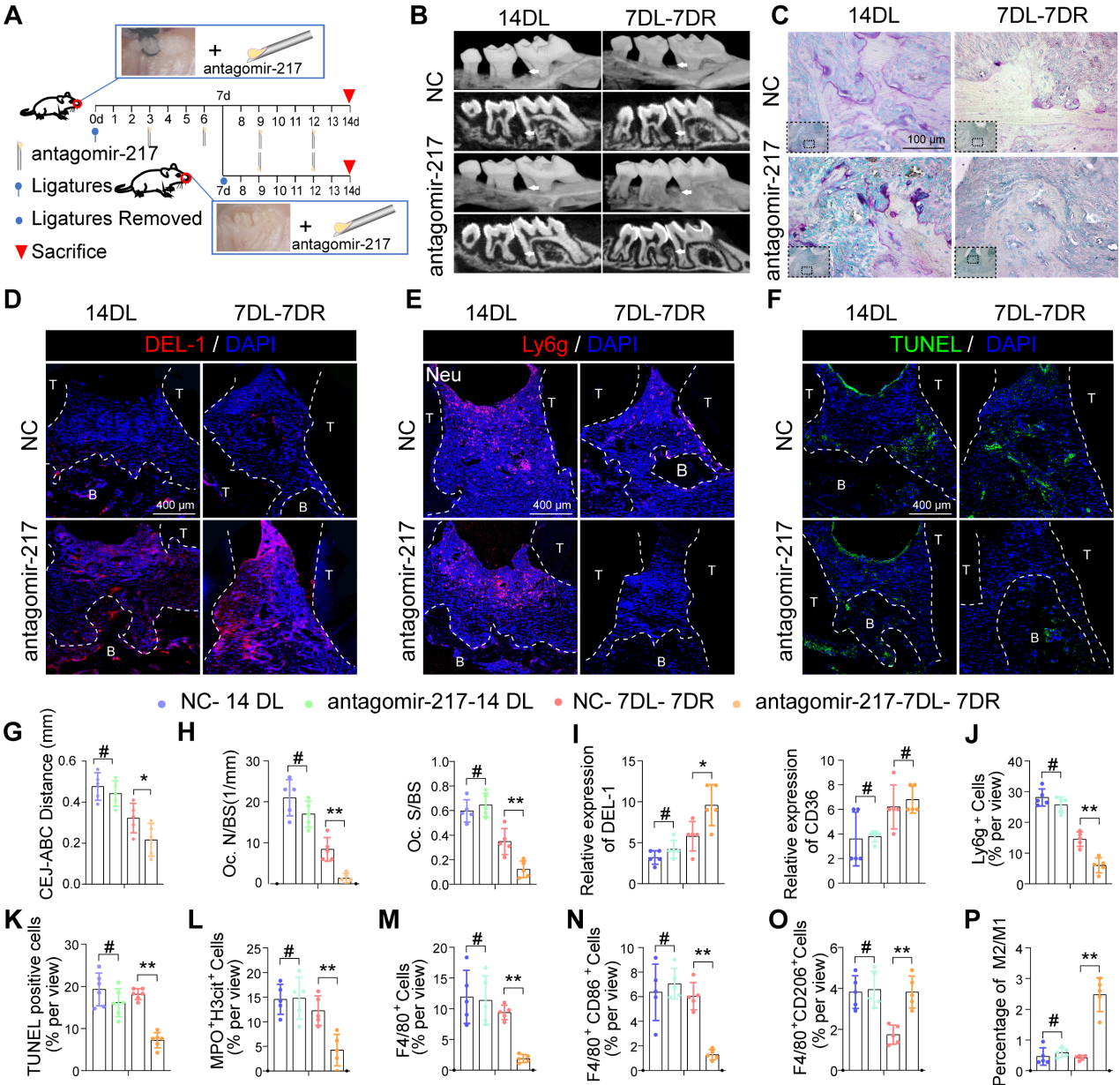
图8 antagomir-217的局部给药促进小鼠DP炎症的消退
结论:
综上所述,作者揭示了SIRT6-miR-216/217轴在糖尿病背景下巨噬细胞胞葬中的重要作用,并概述了通过抑制miR-217来改善炎症消退和牙周组织修复的方法。这一策略可能与糖尿病和其他慢性炎症性疾病的管理相关。
参考文献:
Li B, Xin Z, Gao S, et al. SIRT6-regulated macrophage efferocytosis epigenetically controls inflammation resolution of diabetic periodontitis. Theranostics. 2023 Jan 1;13(1):231-249.