






p53 GOF突变体与ERG通过β-Catenin激活和嘧啶合成促进前列腺癌

TP53肿瘤抑制基因是人类癌症中最常见的突变基因。在约50%的前列腺癌症(PCa)患者中会发生ETS相关基因(ERG)与TMPRSS2基因融合。在本研究中,作者确定p53功能获得(GOF)突变体基因的反式激活功能,并揭示β-Catenin是p53 GOF突变体的转录靶基因,也是TMPRSS2-ERG和p53 GOF突变体阳性PCa驱动和治疗靶点。该研究于2023年8月发表在《nature communications》,IF:16.6。
技术路线













1. 前列腺肿瘤中TMPRSS2- ERG转基因与Trp53缺失协同和p53 GOF突变体的作用
在TCGA数据与转移性PCa SU2C队列数据中,作者确定TMPRSS2-ERG融合与TP53失活在前列腺癌患者中同时发生(图1a,b)。
作者以前列腺特性(Pb)驱动的Cre重组酶转基因小鼠(Pb-Cre4)、Pb-TMPRSS2-ERG(T2-ERG)转基因小鼠、Trp53loxp-top-loxp-R172H/loxp小鼠为基础,共产生六组基因工程小鼠(GEM):(1)野生型(WT)对照;(2) 前列腺细胞特异性T2-ERG转基因(Pb-T2-ERG);(3)前列腺细胞特异性Trp53敲除(Pb-Cre+;Trp53p/p或Trp53pc-/-);(4)前列腺细胞特异性Trp53敲除和R172H突变体敲入(Pb-Cre+;Trp53R172H/p或Trp53pcR172H/-);(5)前列腺细胞特异性T2-ERG转基因和Trp53敲除(Pb-Cre+;Pb-T2-ERG;Trp53p/p或Pb-T2-RG;Trp53pc-/-);(6)前列腺细胞特异性T2-ERG转基因和Trp53敲除/敲入(Pb-Cre+;Pb-T2-ERG;Trp53R172H/p或Pb-T2-RG;Trp53pcR172H/-)(图1c)。TP53 R175H被认为是GOF突变。TMPRSS2-ERG过表达和Trp53缺失诱导10月龄Trp53pc-/-小鼠局灶性低前列腺上皮内瘤变(LGPIN)和约50% 15月龄小鼠高PIN(HGPIN)和局灶性腺癌(图1c,d)。重要的是, PbT2 ERG';Trp53pcR172H/-小鼠在10月龄时发展为HGPIN和腺癌,约90%小鼠在15个月龄时发展为侵袭性HGPIN和/或腺癌(图1c,d)。值得注意的是, IHC结果显示,这些癌症和PIN病变均为雄激素受体(AR)阳性(图1c)。与之前的报道一致,在10月龄Pb-T2-ERG小鼠中未观察到PIN;然而,20%的小鼠在15月龄时出现局灶性LGPIN病变(图1d)。作者所观察到的年龄依赖性疾病发展进一步支持ERG过表达通过与其他基因改变协同促进前列腺肿瘤发生。在Pb-T2-ERG;Trp53pc-/-和Pb-T2-ERG;Trp53pcR172H/-小鼠两个年龄段中,组织学变化与增加的Ki67染色一致。
除R175H突变,在人前列腺癌细胞系VCaP(TMPRSS2-ERG(ERGΔN39)/TP53R248W/-DBD突变体阳性)中,作者证明ERG或p53 DNA结合域(DBD)突变体单独敲除或双敲除抑制VCaP细胞生长(图1g,h)。总之,这些数据表明,小鼠Trp53中R172H和人TP53中R248W等突变分别作为GOF突变驱动小鼠前列腺肿瘤发生和人前列腺癌细胞生长。
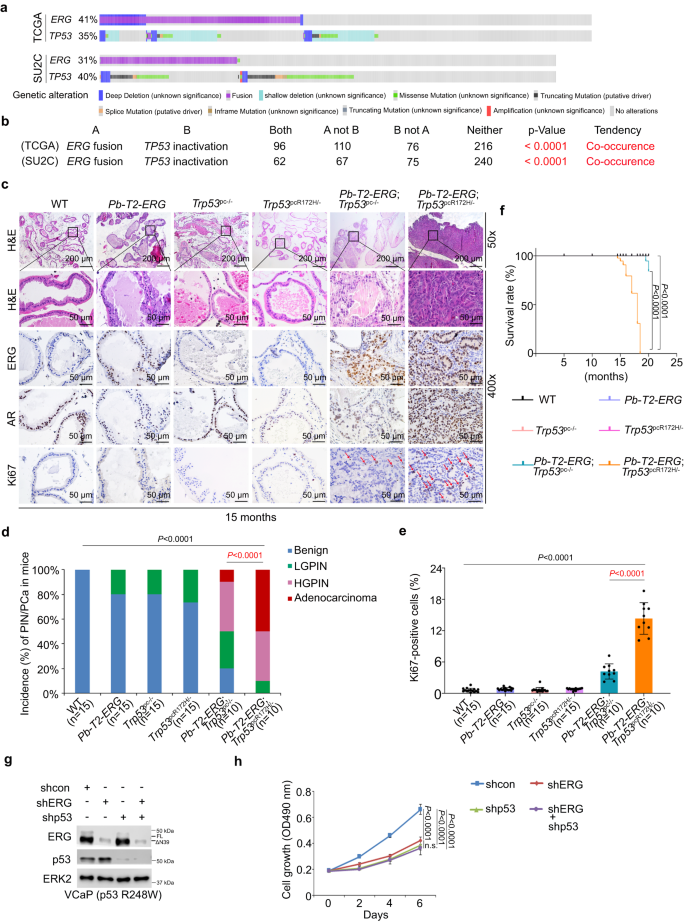
图1:TMPRSS2-ERG融合和TP53失活协同诱导小鼠前列腺肿瘤发生
2. ERC与p53 GOF突变体上调PSGs
作者对六组GEM的前列腺组织进行RNA-seq分析。RNA-seq数据显示,来源于Pb-T2-ERG;Trp53pc-/-小鼠和Pb-T2-ERG;Trp53pcR172H/- PIN病变的前列腺肿瘤共享有370个上调基因,各自有901个和304个独特上调靶点(图2a)。通过对这组基因数据和先前报道的GEM模型中TMPRSS2-ERG前列腺肿瘤产生的ERG ChIP-seq数据整合分析,作者从Pb-T2-ERG;Trp53pcR172H/-小鼠中鉴定出531个前列腺肿瘤中高度上调的ERG靶基因(图2b,c)。IPA分析表明,这些基因中多数与癌症相关(图2d)。与来自对照组小鼠的前列腺组织相比,PSGs亚群,包括Umps、Rrm1、Rrm2 和 Tyms在Pb-T2-ERG;Trp53pcR172H/-肿瘤中上调(图2a,c-g)。这些结果通过Pb-T2-ERG;Trp53pcR172H/-小鼠肿瘤RT-qPCR进一步得到验证(图2h,i)。这些数据表明,p53 GOF突变体与T2-ERG协同上调前列腺癌PSG表达。
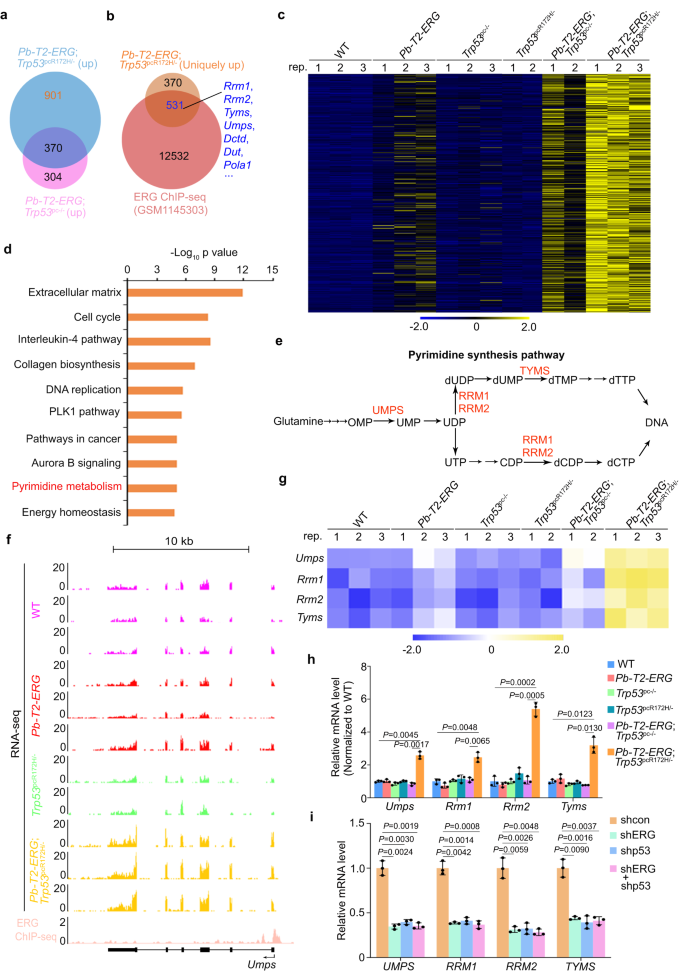
图2:ERG和p53GOF突变体对GEM前列腺肿瘤和人前列腺癌细胞系嘧啶合成基因的协同调控
3. CTNNB1被鉴定为p53 GOF突变体的转录靶基因
为确定p53 GOF突变体是否通过与PSG基因组位点结合来调节PSG表达,作者使用抗p53抗体在VCaP细胞中进行p53 ChIP-seq。共鉴定到1116个与p53 GOF突变体R248W显著结合的峰(P<1E-10),这些峰定位于启动子和非启动子区域,并与615个基因相关(图3a)。但在VCaP细胞中没有检测到p53 GOF突变体R248W与典型p53靶基因的结合,也没有检测到R248W在PSG上的结合位点。这表明p53 GOF突变体间接调控PSG表达。
为明确介导p53 GOF突变体调控PSG表达的下游效应因子,作者对p53突变体R248W结合基因进行GO分析,发现其富集于转录共调节因子结合途径(图3b),并在CTNNBI基因启动子中检测到R248W结合峰,该基因编码β-Catenin (图3c)。在VCaP细胞中通过ChIP-qPCR进一步验证该结果(图3d)。
接下来,作者对p53 R248W ChIP qPCR进行分析(图3e)。他们发现R248W突变体特异性占据VCaP细胞中ChIP-seq峰的中心(图3e,f)。使用VCaP细胞裂解物和生物素标记的双链DNA探针进行电泳迁移率偏移测定(EMSA)确定突变型p53结合序列(MP53BS)(图3e)。在测定中添加竞争性未标记探针或抗p53抗体,MP53BS探针的EMSA信号显著减弱(图3h,i)。另外,作者还证实,p53所有DBD突变体都与MP53BS探针结合(图3j),表明p53 DBD突变体直接与CTNNB1启动子的MP53BS结合。
作者使用CRISPR/Cas9敲除了DU145细胞中CTNNB1启动子MP53BS序列并产生4个MP53BS敲除克隆。在这4个克隆当中,CTNNB1 mRNA和β-Catenin表达均显著下调(图3k,l)。虽然p53 GOF突变体敲除显著降低对照DU145细胞中β-Catenin mRNA和蛋白质表达,但它未能进一步降低MP53BS KO细胞中β-Catenin表达(图3m,n)。这些数据表明β-Catenin是p53 GOF突变体的调节靶点,并通过CTNNB1启动子中MP53BS介导。
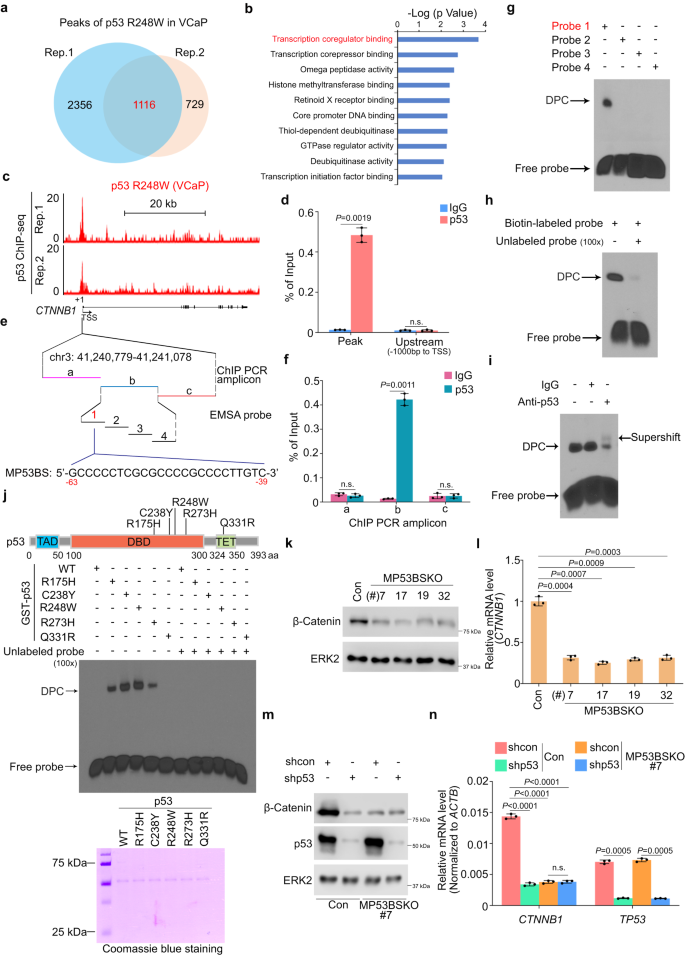
图3:GOF突变体p53与启动子结合并反式激活CTNNB1基因表达
4. TMPRSS2-ERG与p53 GOF突变体协调β-Catenin表达
RNA-seq与RT-qPCR结果显示,Ctnnb1 mRNA在Pb-T2-ERG;Trp53pcR172H/-小鼠前列腺肿瘤中上调(图4a,b,c)。CTNNB1 mRNA仅在SU2C队列的ERG融合和TP53突变双阳性患者样本中显著上调(图4d)。作者不仅发现ERG与CTNNB1启动子结合,而且还鉴定出MP53BS相邻的ERG结合序列(ERGBS)(图4e)。ChIP和re-ChIP证实突变型p53和ERG与VCaP细胞中CTNNB1启动子同一区域结合(图4f)。VCaP细胞中ERG敲低下调CTNNB1 mRNA和β-Catenin蛋白水平(图4g,h),支持GOF突变型p53在调节PCa细胞β-Catenin表达中的主要作用。通过敲低VCaP细胞中ERG或p53突变体,降低了H3K27Ac(基因转录的活性标记物)在CTNNB1启动子处的富集(图4i)。萤光素酶报告基因分析结果显示,CTNNB1启动子活性通过不同的p53 GOF突变体单独表达而增加,而不是通过单独表达ERG;然而,p53突变体和ERG共表达显著增强启动子活性(图4j)。这些数据表明TMPRSS2-ERG与p53 GOF突变体协同调节PCa细胞中CTNNB1表达。
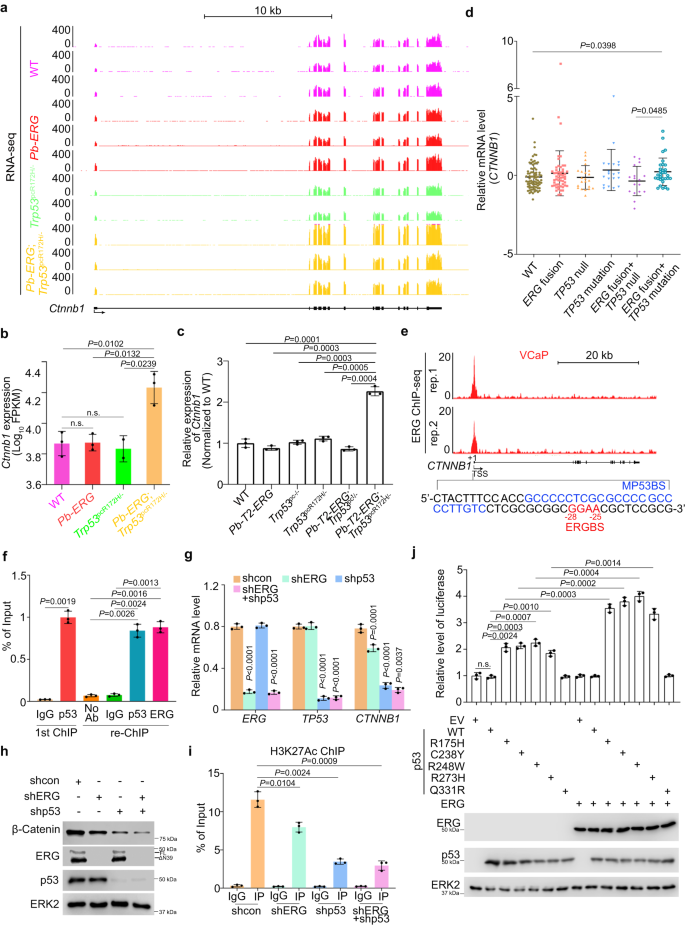
图4:ERG和GOF突变型p53共调节β-Catenin表达
5. TMPRSS2-ERG与β-Catenin协调PSG表达
β-Catenin与PSG的启动子和/或非启动子区域结合,包括UMPS、RRM1、RRM2和TYMS(图5a)。ChIP-qPCR进一步证实ERG和β-Catenin在这些基因座的启动子或增强子上的存在(图5b,c)。
β-Catenin单独敲低抑制VCaP细胞中PSG mRNA和蛋白表达(图5d,e),ERG或p53 R248W敲低则不会导致PSG mRNA和蛋白在β-Catenin缺失细胞系中进一步降低表达(图5d,e)。
作者敲低VCaP细胞中内源性ERGΔN39和p53突变体R248W,并测定嘧啶合成的三个关键中间体UMP、dTTP和dTDP水平(图2e)。UMPS、RRM1和RRM2这三种嘧啶合成的关键酶单独或同时耗竭(图2e)在很大程度上抑制VCaP细胞生长(图5i,j)。同时,作者证明Dox诱导的PSG敲低在很大程度上抑制了小鼠中VCaP肿瘤生长(图5k,l)。Dox给药也降低了肿瘤重量(图5m)。这些数据表明,ERG和p53突变体诱导上调关键的PSG,如UMPS、RRM1和RRM2,这对于体外体内TMPRSS2-ERG和p53突变体双阳性PCa细胞的生长很重要。

图5:ERG与β-Catenin共占据PSG位点并共调控其表达
6. β-Catenin抑制剂在体内体外抑制PSG表达与TMPRSS2-ERG/ p53突变体阳性PCa细胞生长
ICG-001与PRI-724是β-Catenin抑制剂。这两种药物在p53突变体R248W阳性VCaP细胞中表现出更强的生长抑制作用(图6a,b)。ICG-001处理VCaP细胞降低了PSG 表达,以及β-Catenin靶基因CCND1和c-MYC mRNA和蛋白水平的表达,并以剂量依赖性方式抑制细胞生长(图6c–e)。用PRI-724处理VCaP细胞也获得了类似的结果(图6f–h)。接下来,作者在体内检测了β-Catenin抑制剂ICG-001的抗癌作用。ICG-001给药显著抑制了小鼠VCaP异种移植物肿瘤生长(图6i–k)。IHC显示,ICG-001处理降低了嘧啶合成酶如UMPS、RRM1和Ki67的表达,但没有降低CREB结合蛋白(CBP)表达(图6l,m)。
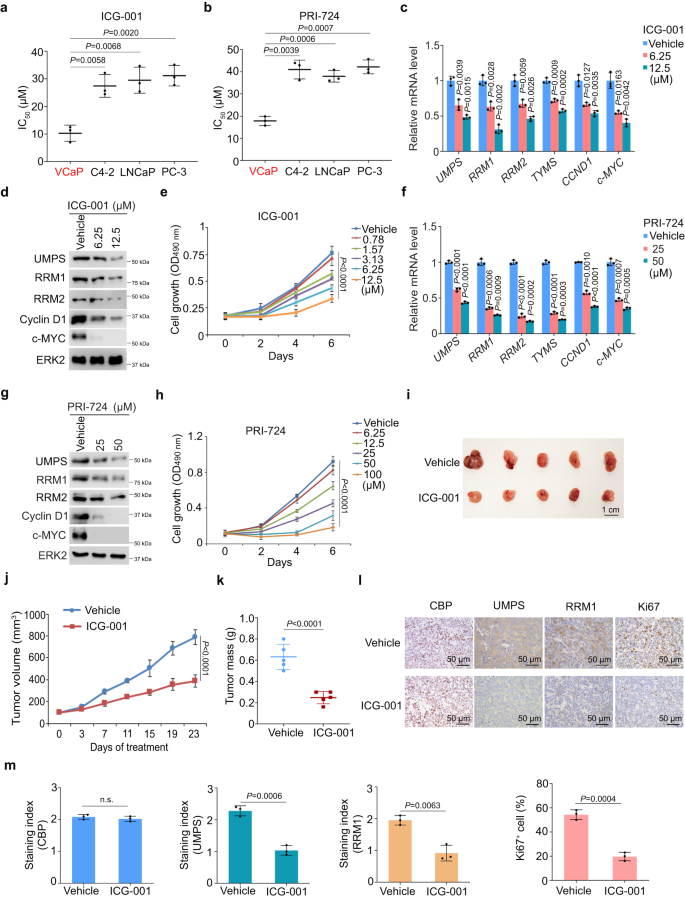
图6:β-Catenin抑制剂抑制PSG表达和小鼠PCa异种移植物生长
7. β-Catenin-LEF/TCF是TMPRSS2-ERG/ p53突变体阳性
β-Catenin通过与DNA结合伴侣LEF/TCF家族蛋白(包括LEF1、TCF1、TCF3和TCF453)形成蛋白质复合物来反式激活靶基因。作者最近报道基于寡核苷酸的PROTACs(O’PROTACs或OP)开发,以靶向LEF1进行蛋白质破坏。有效的LEF1 O’PROTAC(OP-V1)几乎完全溶解VCaP细胞中LEF1蛋白。这种O’PROTAC也在一定程度上下调TCF3和TCF4蛋白,这与LEF/TCF蛋白家族成员共享相似核心DNA靶序列(例如TCAAAG)观察结果一致(图7a,b)。重要的是,这种LEF1/TCF OP还抑制关键嘧啶合成酶蛋白质表达和培养中VCaP细胞生长(图7b,c)。据报道,LuCaP 23.1 PDX及其雄激素依赖(去势抗性)亚系LuCaP 23.1AI是TMPRSS2-ERG基因融合阳性,并且TP53缺失一个等位基因。与LuCaP 23.1AI报道一致,作者证实亲代LuCaP 23.1-PDX肿瘤在p53 DBD中也存在C238Y突变(图7d)。突变型p53 C238Y与CTNNB1启动子MP53BS结合(图3j),shRNA敲低该突变型可显著降低LuCaP 23.1 PDX衍生的类器官(PDXO)中β-Catenin表达(图7e)。
作者证明,LEF1/TCF OP处理不仅抑制关键嘧啶合成酶蛋白表达,而且有抑制LuCaP 23.1 PDXO生长(图7f–h)。补充dTTP/dCTP几乎完全逆转这种作用,但补充dATP/dGTP脱氧核苷三磷酸盐没有显著效果(图7g,h)。LEF1/TCF OP处理显著阻碍LuCaP 23.1 PDX肿瘤的生长,而不会导致小鼠体重发生降低(图7i–l)。ILEF1/TCF-OP不仅降低LEF/TCF蛋白和嘧啶合成酶如UMPS和RRM1表达水平,而且减少了Ki67阳性细胞数量(图7m,n)。这些结果表明,O’PROTAC通过靶向LEF/TCF蛋白抑制β-Catenin和PSG表达,体内外有效阻断了TMPRSS2-ERG和GOF-p53突变阳性PCa生长。

图7:LEF1/TCF O 'PROTAC抑制PSG表达和TMPRSS2- ERG和突变型p53阳性PCa PDX肿瘤生长
结论
在本研究中,作者确定p53 GOF突变体基因的反式激活功能,并揭示β-Catenin是p53 GOF突变体的转录靶基因,也是TMPRSS2-ERG和p53 GOF突变体阳性PCa驱动和治疗靶点。
实验方法
IHC,CRISPR/Cas9,RT-qPCR,Western blot,RNA-seq
参考文献
Ding D, Blee AM, Zhang J, Pan Y, Becker NA, Maher LJ 3rd, Jimenez R, Wang L, Huang H. Gain-of-function mutant p53 together with ERG proto-oncogene drive prostate cancer by beta-catenin activation and pyrimidine synthesis. Nat Commun. 2023 Aug 3;14(1):4671. doi: 10.1038/s41467-023-40352-4. PMID: 37537199.