






中和IL-8可以增强胶质瘤ICB疗效
恶性神经胶质瘤预后很差,由于标准治疗的选择有限,仍然具有挑战性。尽管免疫检查点阻断(ICB)疗法在治疗多种肿瘤方面取得了显著的成功,但ICB疗法治疗恶性胶质瘤的临床益处仍然有限。因此探究联合靶点是十分必要的,该文于2023年4月发表在《Cancer Cell》,IF=50.3。
技术路线

主要研究结果
1、胶质瘤患者肿瘤浸润T细胞的解剖分析
作者测量了肿瘤中的T细胞驻留模式和转录组,胶质瘤组织和外周血样本上的表面标记和转录组,样本包括51名低级别胶质瘤(LGG)患者和101名高级别胶质瘤(HGG)患者(图1A)。作者首先用H&E和免疫组织化学(IHC)对71例胶质瘤患者的肿瘤组织进行染色,包括HGG和LGG。值得注意的是,大多数T细胞位于微血管区的血管周围 (图1B),解剖转录图谱数据证实了这一点(图1C)。为了弄清是哪种趋化因子受体引导T细胞浸润到胶质瘤中,作者利用CyTOF检测了14名新诊断患者的外周血单个核细胞(PBMCs)和胶质瘤组织样本,其中包括5对PBMCs和组织样本。作者的研究结果显示,在所检测的趋化因子受体中,包括CCR4、CCR5、CCR6、CXCR3和CX3CR1,与外周血相比,只有CCR5和CXCR3在肿瘤CD4+和CD8+ T细胞上的表达均显著升高(图1D),这表明需要CCR5和CXCR3来引导T细胞向胶质瘤迁移。
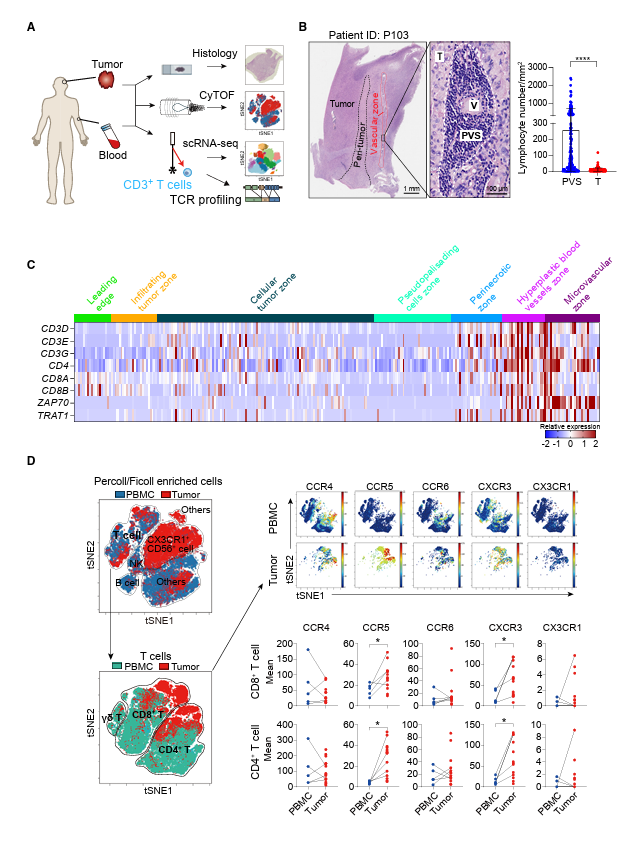
图1. 胶质瘤中T细胞的景观定位和解剖分析
2、胶质瘤中T细胞单细胞测序
为了全面剖析肿瘤微环境(TME)中T细胞的异质性,作者从5个新分离的肿瘤和配对的PBMC中分离T细胞,并通过scRNA-seq和单细胞T细胞受体(TCR)测序(scTCR-seq)对它们进行了分析。经过严格的质量控制筛选,作者总共获得了116,205个T细胞(65,990个来自PBMC, 50,215个来自肿瘤)。TCR全库测序鉴定出67,665个具有配对a链和b链的T细胞。利用免疫表位数据库和分析资源(IEDB)工具排除非肿瘤特异性TCR后,剩余的潜在肿瘤反应性TCR克隆型定义为至少两个T细胞共享的配对a链和b链。根据先前的报道,T细胞簇根据其居住地、TCR克隆性和特征基因进行分类。在肿瘤和PBMC样本中共鉴定出18个T细胞簇,包括7个CD4+ T细胞簇,10个CD8+ T细胞簇和1个gd T细胞簇,每个T细胞簇都具有独特的特征基因(图2A)。具体而言,CD4+ T细胞区室多样化,有7个簇。幼稚亚群(C1_CD4_Naive)在血液中占主导地位(图2B)。中枢记忆亚群(C2_CD4_Tcm)在血液中普遍存在,其特征是IL7R、TCF7、SELL和MYC基因的高表达(图2B)。克隆扩增的细胞毒性亚群(C6_CD4_Cyto)细胞高度表达细胞毒性相关基因NKG7、GZMA和GZMK(图2B-2D)。一个功能失调的亚群(C7_CD4_Dysfun)也被克隆扩增,并以编码抑制CTLA-4的基因高表达和最低水平的细胞毒性相关基因为标志(图2C, 2D)。值得注意的是,一个产生IL-8的亚群(C8_CD4_IL8)只在肿瘤中富集,并以IL8、NINJ1、HLA-DRA和HAVCR2高表达为特征(图2B)。另一个非克隆扩增的T细胞亚群(C9_CD4_Tn-like)高度表达幼稚标记基因和SOCS3(图2B),因此代表了先前报道的幼稚样T细胞亚群表达调节性T细胞亚群(C12_Treg)的foxp3被克隆扩增,表明TCR依赖性激活或分化(图2C、2D)。同样,转录特征和TCR克隆扩增的组合分析显示CD8+ T细胞有10个主要集群(图2A),高度克隆扩增的T细胞(TCR克隆大小大于10,TCS > 10)主要位于TME(图2B-2D)。预衰竭CD8+ T细胞(C3_CD8_Pre-Ex, TCS > 10, 37.8%)表达效应基因NKG7、GZMA、GZMH和GZMK以及表面标记基因LAG3、KLRB1、TIGIT和PDCD1,与之前的鉴定一致,优先位于肿瘤部位(图2B)。值得注意的是,效应/记忆CD8+ T细胞在pbmc中优先富集(图2B)。其余4个非克隆或低克隆扩增的CD8+ T细胞簇包括幼稚T细胞簇(C14_CD8_Naive)、中枢记忆亚群(C16_CD8_Tcm)、产生IL-8亚群(C18_CD8_ IL-8)和MGP基因标记的未知簇(C10_ CD8_MGP)。为了探索这6个高度克隆扩增的CD8+ T细胞簇的发育状态和个体发生关系,作者进行了时间轨迹分析和TCR克隆型评估。作者发现,在CD8+ T细胞总数的背景下,这六种不同的集群形成了发育顺序,效应/记忆CD8+ T集群停留在假时间的早期,其次是效应、预衰竭、应激、增殖和衰竭集群(图2E)。基于TCR克隆型共享度的克隆分析也独立验证了这种发育转变。引人注目的是,三种最常见的肿瘤富集CD8+ T细胞亚群,预衰竭,衰竭和效应亚群,构成了大多数克隆T细胞,并且广泛地共享它们的克隆(图2C),表明它们可能是杀死胶质瘤肿瘤细胞的主要局部参与者,并且仍然是胶质瘤的主要免疫治疗靶点。为了进一步验证作者关于趋化因子受体CCR5和CXCR3选择性标记胶质瘤浸润T细胞的观察结果(图1D),总的来说,作者的发现强调趋化因子受体CXCR3和CCR5是T细胞迁移到胶质瘤并执行肿瘤杀伤功能所必需的。
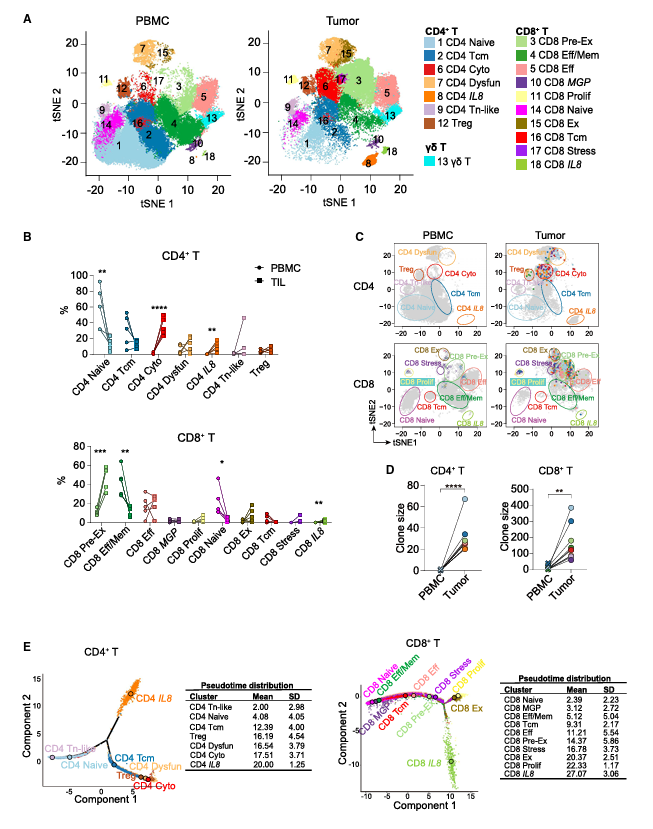
图2. T细胞的单细胞聚集
3、一种独特的产生IL-8的CD4+ T细胞群存在于胶质瘤TME中,预示着不良的临床结果
作者上面的T细胞图谱已经鉴定出一种独特的CD4+ T亚型,它被标记为选择性表达IL- 8,并且只在肿瘤中富集(图3A)。进一步关注CD4+ T细胞区室的转录组评估显示,表达IL-8的CD4+ T细胞表现出先天样特征,大量表达经典巨噬细胞相关效应基因,如IL8、IL1B、CCL3 (MIP-1a)、CCL4 (MIP-1b)和骨髓表面标记基因NINJ1和CX3CR1(图3B)。然后,作者通过免疫组化、免疫荧光(IF)染色和流式细胞术评估了IL-8、CX3CR1和NINJ1蛋白的表达,发现IL-8蛋白在胶质瘤微血管区周围的一部分CD4+ T细胞中高表达,与一般T细胞分布模式一致,这些产生IL-8的CD4+ T细胞也高水平表达CXCR3和NINJ1(图3C)。此外,作者对来自健康供体和胶质瘤患者的75个血液和73个胶质瘤样本的T细胞进行了流式细胞术分析,观察到IL-8在肿瘤样本中活化的CXCR5+ CD4+ T细胞中选择性表达(图3D)。尽管如此,血液中表达IL-8的T细胞优先局限于CXCR5 CD4+ T细胞群(图3D),表明失活或初始CD4+ T细胞。接下来,作者研究了产生IL-8的T细胞簇的免疫特性与临床结果的相关性。首先,作者描述了关键的特征基因表达水平,并将其指定为特征分数。在两种GBM中,产生IL-8的CD4+ T细胞的特征评分越高,总生存率越差(风险比[HR][95%可信区间(CI)] = 4.09 [2.68, 6.25];p < 0.0001)和LGG患者(HR [95% CI] = 2.98 [2.02, 4.39];p < 0.0001)(图3E)。IL-8在胶质瘤中的表达与其受体CXCR1和CXCR2均呈正相关,可独立预测胶质瘤患者预后不良(图3F)。总的来说,作者的研究证明了一种独特的产生IL-8的CD4+ T细胞群,这与胶质瘤患者的低生存率有关。
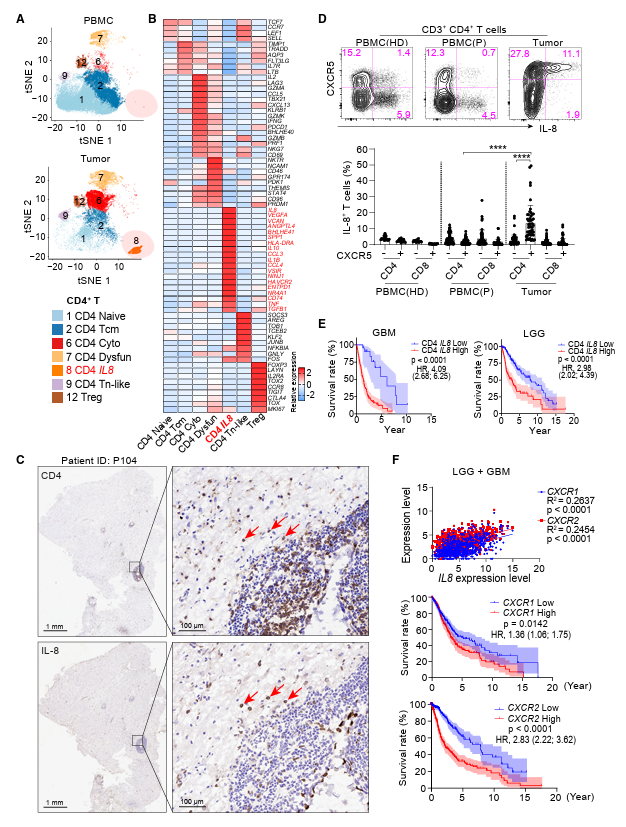
图3. 产生IL -8的CD4+ T细胞亚群预示着不良的临床预后
4、由肿瘤分泌刺激分化的产生IL-8的T细胞表现出先天样的特征,具有促进肿瘤生长和骨髓白细胞募集的能力
胶质瘤浸润T细胞的伪时间轨迹分析显示,IL-8的表达仅限于终末分化和非克隆CD4+ T细胞(图2E),这与IL-8在幼稚CD4+ T细胞(尤其是儿童或脐带血)中的自发表达形成对比。作者假设胶质瘤中产生IL-8的T细胞被肿瘤因子极化,并且独立于TCR信号传导。为了验证这一假设,作者从健康供体中分离出人类原始CD4+ T细胞,并用胶质瘤培养上清液刺激它们。IL-8在T细胞中的表达以时间和剂量依赖的方式升高至 55%(图4A)。基于大量RNA测序数据的分层聚类分析显示,体外分化的产生IL-8的T细胞具有与体内CD4_IL8集群相似的基因表达模式(图4B)。具体来说,2064个基因的表达(增加1487个;down, 577)在体外极化产生IL-8的T细胞和C8_CD4_IL8簇中都发生了相同的变化(图4B)。通过基因集富集分析(GSEA)和创新途径分析(IPA)进一步评估超过2倍改变的基因,发现产生IL-8的T细胞上调巨噬相关基因,包括IL-8、IL-1B、IL-6、CCL2、CCL3、CCL4、CCL20、CHI3L1、CX3CR1等,具有类似于胶质瘤相关骨髓细胞的先天分子特征(图4C和4D),可能在功能上与血管生成、单核细胞/中性粒细胞趋化性、和氧化还原。这些结果共同强调了先天样IL-8+ CD4+ T细胞作为一种独特的CD4+ T效应谱系。接下来,作者试图确定产生IL-8的CD4+ T细胞在胶质瘤中的功能。作者分别以1:1的比例将U373细胞接种到免疫缺陷小鼠的中枢神经系统中,或与幼稚和体外生成的产生IL-8的CD4+ T细胞一起接种。接受IL-8+ CD4+ T细胞的小鼠与初代T细胞小鼠和对照组相比,肿瘤生长明显(图4E)。肿瘤浸润免疫细胞的离体分析显示,与其他两组相比,IL-8+ CD4+ T接受组CD45+ CD11b+ Gr1+髓样细胞显著增加,供体IL-8+ CD4+ T细胞组成性高表达IL-8(图4F和4G)。为了进一步验证产生IL-8的T细胞功能,作者采用另一种胶质瘤异种移植小鼠模型,将20,000个GBM5细胞(作者最近研究中描述的一种原发性胶质母细胞瘤细胞系)接种到免疫功能低下的小鼠脑内(图4H)。如图4I所示,供体产生IL-8的T细胞与原位骨髓细胞紧密结合,与对照组相比吸引了更多的骨髓细胞。这些结果共同强调了IL-8+ CD4+ T细胞在肿瘤中的促肿瘤功能、对髓系白细胞的趋化性和促血管生成。鉴于上述观察转录因子BHLHE41起到了足够的作用在偏振引发+ CD4 + T细胞,作者下一个试图评估其必要的角色在这些T细胞,发现消融的BHLHE41 CRISPRCas9系统无法消除引发表达CD4 + T细胞,在人类和自适应传输BHLHE41-knockout CD4 + T细胞没有逆转肿瘤的生长和鼠标生存相比,对照组的争夺(数据没有显示)。因此,这些结果表明BHLHE41在编程表达IL-8的CD4+ T细胞中发挥了充分而非必要的作用。
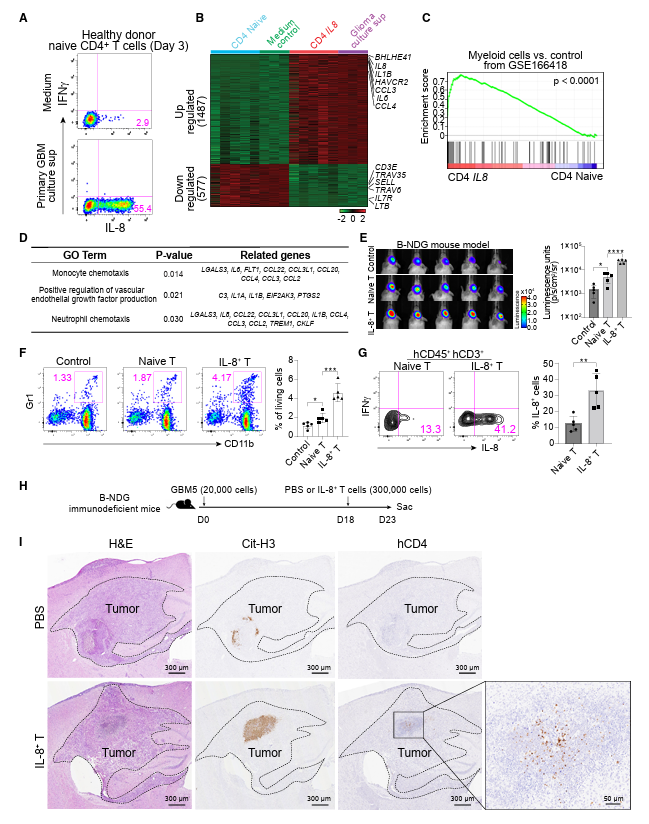
图4. 产生IL -8的T细胞的转录和功能鉴定
5、恶性胶质瘤中主要由肿瘤细胞、髓细胞和T细胞表达的IL-8增强了血管免疫微环境的抑制特性
越来越多的临床证据表明,血浆中IL-8的表达水平与肿瘤负荷呈正相关,但与ICB治疗的临床结果呈负相关,这导致了针对IL-8和/或其受体CXCR1/CXCR2的潜在癌症治疗方向。然而,由于啮齿动物缺乏IL-8基因,IL-8在体内的功能,特别是作为肿瘤发生的关键免疫调节因子,尚不清楚。作者首先研究了恶性胶质瘤中IL-8产生的细胞来源。对人GBM的snRNA-seq数据分析显示,IL8的表达主要局限于肿瘤细胞、髓细胞和T细胞(图5A)。此外,作者遵循之前的策略,生成了IL-8人源化(IL8- hu)小鼠品系,其中将含有整个人类IL-8基因位点和上下游调控元件的~160 kb人类BAC克隆(RPL11-997L11)插入小鼠9号染色体的基因间区(图5B)。与先前研究的观察结果一致,在外观、体重、寿命、生育能力和淋巴器官免疫细胞群方面,幼稚的IL8-Hu小鼠与WT对照小鼠没有区别(数据未显示)。然后,作者将表达荧光素酶的GL261细胞注射到小鼠颅内,以模拟人类胶质母细胞瘤。与WT组相比,移植GL261的IL8-Hu小鼠肿瘤大小明显增大,寿命明显缩短(图5C)。通过IHC和H&E染色对IL8-Hu小鼠肿瘤组织进行组织学检查,发现肿瘤坏死区域和血管密度增加,且常伴有IL-8阳性多形核白细胞(PMN)浸润,其模式与人GBM肿瘤组织非常相似(图5D)。更重要的是,IL-8在IL-8- hu小鼠肿瘤中CD45+ CD3骨髓细胞和CD4+ T细胞中的表达显著升高,这些小鼠IL-8+ CD4+ T细胞转录反映了人类表达IL-8的CD4+ T细胞(图5E)。此外,与对照组相比,IL8-Hu小鼠的肿瘤浸润性CD4+和CD8+ T细胞携带更多的衰竭标志物PD-1和TIM-3(图5F)。因此,与WT小鼠相比,作者新生成的IL8-Hu小鼠品系更适合模拟人类胶质瘤,在胶质瘤中,人IL-8的生理表达能够再现肿瘤生长加速和血管生成,并增强骨髓源性抑制细胞在TME中的募集。

图5. IL-8人源化小鼠品系能够更好地模拟胶质瘤患者的TME
6、阻断IL-8-CXCR1/CXCR2轴与抗pd -1免疫治疗协同作用
作者研究了阻断IL-8-CXCR1/ CXCR2轴是否能增强抗pd -1在恶性胶质瘤中的作用。药物抑制剂reixin抑制CXCR1和CXCR2,可显著减缓IL8-Hu和WT小鼠的肿瘤生长,提高生存率(图6A和6B)。流式细胞术评估显示,修复素治疗不仅消除了抗pd -1诱导的CD11bhiGr1hi MDSCs和CD11bhiGr1mid MDSCs的基础水平,也消除了肿瘤中CD11bhiGr1hi MDSCs和CD11bhiGr1mid MDSCs的基础水平,但在IL8-Hu小鼠和WT小鼠的脾脏中没有(图6C、6D)。重要的是,PD-1的阻断与修复素一起,进一步将Tim-3和PD-1在T细胞中的表达降低到最低水平,尽管浸润肿瘤的T细胞总数没有显著变化(图6E)。因此,作者的研究结果为以下观点提供了证据:通过靶向CXCR1和CXCR2隔离MDSCs可以利用抗pd -1治疗胶质瘤的疗效。接下来,作者通过检查IL-8的中和是否具有类似的效果来扩展作者的研究。由于人原发性胶质母细胞瘤细胞也高水平表达IL-8,作者通过将IL-8和荧光素酶双表达的GL261细胞注射到IL8-Hu小鼠中,然后用PD-1和/或IL-8阻断抗体对其进行顺序处理,严格模拟人胶质母细胞瘤(图6F)。接受抗IL-8抗体的小鼠肿瘤大小明显减小,生存率明显提高(图6G和6H)。值得注意的是,与同型对照组相比,PD-1和IL-8的双重阻断显著延长了中位生存期18天(图6H)。鉴于作者之前的数据显示抗pd -1抗体在IL8-Hu小鼠中增加了全身IL-8,并且IL-8能够促进肿瘤血管生成,作者通过CD31免疫组化染色对胶质瘤肿瘤的微血管进行了组织学评估。与对照小鼠相比,接受抗pd -1抗体治疗的荷瘤小鼠IL-8 - hu微血管密度显著升高,而抗IL-8治疗后微血管密度大大降低(图6I)。总之,作者的研究结果表明,IL-8的中和相当于其受体CXCR1和CXCR2的阻断,通过消除抑制性MDSCs来增强T细胞介导的抗肿瘤免疫。
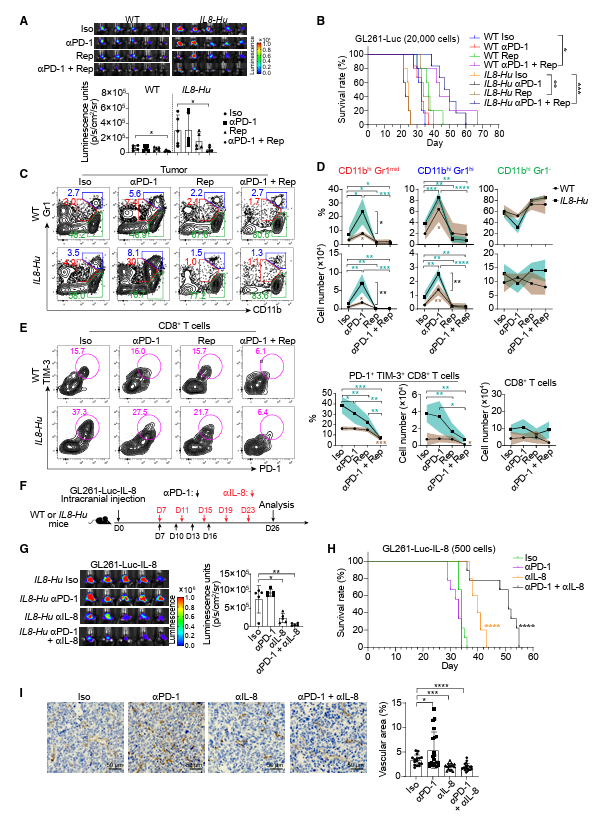
图6. 阻断IL-8-CXCR1/CXCR2轴可发挥抗PD-1的作用
7、阻断IL-8重编程胶质瘤肿瘤微环境
髓系细胞,包括单核细胞来源的巨噬细胞(MDMs)、小胶质细胞和MDSCs,构成胶质瘤TME的主要非肿瘤性细胞成分。41,42接下来,作者进行了scRNA-seq分析,以探讨在单独阻断PD-1和IL-8以及联合阻断的情况下胶质瘤TME的变化。如图7A-7C所示,对肿瘤中CD45+细胞的scRNA-seq数据进行无监督聚类分析,鉴定出10种细胞亚型,包括小胶质细胞、T细胞、MDM、M-MDSC、G-MDSC、树突状细胞(DC)、活化DC、浆细胞样DC (pDC)、肥大细胞和B细胞。与作者流式细胞术观察的结果一致,与WT小鼠相比,更多的MDSCs浸润到IL8-Hu的TME中,抗pd -1治疗进一步特异性增强了M-MDSCs的浸润,而IL-8中和消除了肿瘤浸润的M-MDSCs,无论是否存在抗pd -1抗体(图7D和7E)。此外,研究发现M-MDSCs容易产生活性氧,例如脂肪酸氧化相关基因Alox12e、Alox15和Pla2g3的选择性表达,以及超氧阴离子产生相关基因Prg3、Tafa4和Nox1(图7F),这突出了G-MDSCs和M-MDSCs在协调肿瘤免疫抑制微环境方面的不同作用(TIME)。此外,据报道,促进TIME的肿瘤浸润性肥大细胞几乎被IL-8中和耗尽(图7D)。肿瘤相关巨噬细胞(tam),包括MDMs和小胶质细胞,在WT和IL8-Hu小鼠的胶质瘤TME中都大量富集(图7D)。为了进一步探索IL-8中和对tam的潜在影响,作者将MDM集群重新划分为四个亚集群,包括亚集群1(2型样巨噬细胞,m2样)、亚集群2(抗肿瘤)、亚集群3(抗血管生成)和亚集群4(图7G和7H)。作者发现,接受非抗IL-8治疗的小鼠的肿瘤浸润性MDM细胞大部分(80%)由具有免疫抑制和血管生成相关基因的m2样巨噬细胞组成(图7H和7I)。相比之下,接受抗IL-8单药或联合治疗的小鼠MDM细胞主要由亚群3、抗血管生成巨噬细胞组成(图7H、7I)。因此,作者的研究结果表明,IL-8的中和通过将m2样巨噬细胞转换为抗血管生成巨噬细胞来重新编程胶质瘤中的MDMs。同样,胶质瘤驻地小胶质细胞被重新分为四种亚型,包括静息小胶质细胞、抑制性小胶质细胞、IL-13+细胞和炎性小胶质细胞(图7J和7K)。最引人注目的是,IL-8抑制有效地消融了具有免疫抑制和血管生成相关基因的抑制性小胶质细胞(图7I)。综上所述,作者的上述数据表明,IL-8阻断可以通过消除MDSCs和肥大细胞以及tam的重新填充,将胶质瘤TME从促肿瘤状态重塑为抗肿瘤状态,从而与ICB治疗协同产生整体抗肿瘤免疫反应。
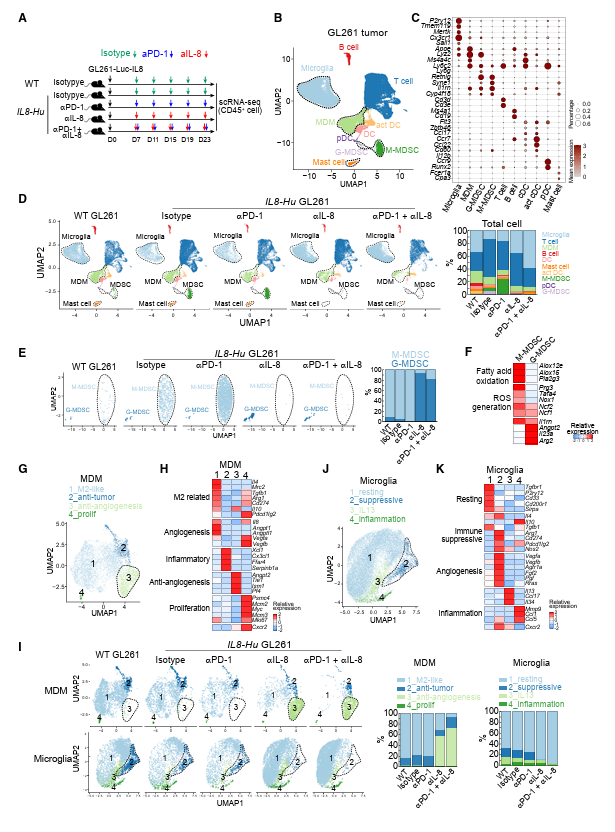
图7. 阻断IL-8可重编程肿瘤免疫微环境
结论
总之,作者定义了一个治疗上可靶向的趋化轴,并且靶向IL-8/CXCR1/ CXCR2轴联合ICB可能使胶质瘤患者受益。
实验方法
细胞悬浮液的制备、scRNA-seq、血细胞计数、T细胞体外分化、流式细胞术细胞表面和细胞内细胞因子染色、免疫荧光(IF)和免疫组化染色、ChIP-qPCR、ELISA、RNA测序、血管形成实验
参考文献
Liu H, Zhao Q, Tan L, Wu X, Huang R, Zuo Y, Chen L, Yang J, Zhang ZX, Ruan W, Wu J, He F, Fang Y, Mao F, Zhang P, Zhang X, Yin P, Yan Z, Xu W, Lu H, Li Q, Liang M, Jia Y, Chen C, Xu S, Shi Y, Ping YF, Duan GJ, Yao XH, Han Z, Pang T, Cui Y, Zhang X, Zhu B, Qi C, Wang Y, Lv SQ, Bian XW, Liu X. Neutralizing IL-8 potentiates immune checkpoint blockade efficacy for glioma. Cancer Cell. 2023 Apr 10;41(4):693-710.e8. doi: 10.1016/j.ccell.2023.03.004. Epub 2023 Mar 23. PMID: 36963400.