






己糖激酶2通过组蛋白乳酸化促进肝纤维化
肝纤维化是肝脏对各种损伤的伤口愈合反应,与严重的发病率和死亡率有关。目前,除肝移植外,尚无治疗肝纤维化的有效方法。在肝损伤期间,静止的肝星状细胞(HSC)被激活并转分化为α -平滑肌肌动蛋白(a-SMA)阳性的肌成纤维细胞,这是纤维化肝脏中胶原生成细胞的主要贡献者。最近,包括有氧糖酵解在内的代谢重编程已成为HSC激活的一个关键特征,有证据表明,抑制有氧糖酵解可阻止HSC激活。尽管观察到在HSC激活过程中有氧糖酵解的增强会增加乳酸的产生,但乳酸与HSC激活相关的潜在机制仍然难以捉摸。该文章就该问题进行了详细的阐述,于2023年9月发表在《Cell Metabolism》,IF=29。
技术路线

主要研究结果
1、HK2的催化活性诱导组蛋白乙酰化,但不诱导组蛋白乙酰化
为了探讨HK2的表达及其催化活性是否影响组蛋白乳酸化,作者使用了中国仓鼠卵巢(CHO)细胞MI5-4(HK基因表达不可见且HK活性有限的)。作者在这些细胞中表达野生型(WT) HK2或激酶死亡突变型(SA) HK2。在突变体中,丙氨酸取代了催化活性所需的氨基末端和羧基末端的丝氨酸残基(S155A/S603A)。如图1A所示,在MI5-4 CHO细胞中,WT和SA HK2的表达水平相同。然而,细胞外酸化率(ECAR)测量显示,糖酵解活性仅在WT HK2细胞中显著增加(图1B和1C)。WT HK2细胞的糖酵解活性也导致乳酸产生和细胞乳酸水平的显著诱导(图1D)。为了确定HK2诱导的乳酸生成是否足以促进细胞中组蛋白的乳酸化,作者检测了H3K18la。赖氨酸残基也被乙酰化(H3K18ac)。有趣的是,H3K18la在空载体(EV)和SA HK2细胞中几乎检测不到,但在WT HK2细胞中却高度升高,而H3K18ac无论HK2表达如何都可以检测到,在WT HK2细胞中略有降低(图1E)。除了H3K18la外,作者发现H3K9la、H3K14la、H4K8la和H4K12la也仅在WT HK2细胞中升高。作者还观察到,在WT HK2细胞中,非组蛋白的乙酰化程度大多升高,而在WT HK2细胞中,尽管细胞乙酰辅酶a水平较高,但大多数非组蛋白的乙酰化程度没有增加,甚至降低。为了进一步阐明H3K18la和H3K18ac的潜在功能意义,作者接下来进行了一种新的全基因组免疫检测CUT&Tag。对H3K18la和H3K18ac的全基因组分布分析表明,这两种组蛋白修饰主要位于启动子区域(图1F)。与作者的免疫印迹实验结果一致,作者还观察到,在WT HK2中,H3K18la峰在转录起始位点(tss)附近增加,但H3K18ac峰没有增加H3K18ac峰甚至略有下降(图1G)。为了确定H3K18la和H3K18ac调控的潜在候选基因,作者首先比较了相同基因启动子区域的组蛋白修饰水平。在WT HK2细胞中,大多数基因标记为H3K18la升高,而H3K18ac未被显著诱导(图1H和1I)。只有在WT HK2细胞中,这些基因的H3K18ac均未升高。因此,虽然HK2的表达,通过增加糖酵解,可以增加乳酸和乙酰辅酶a的水平,但它只影响组蛋白的乳酸化。
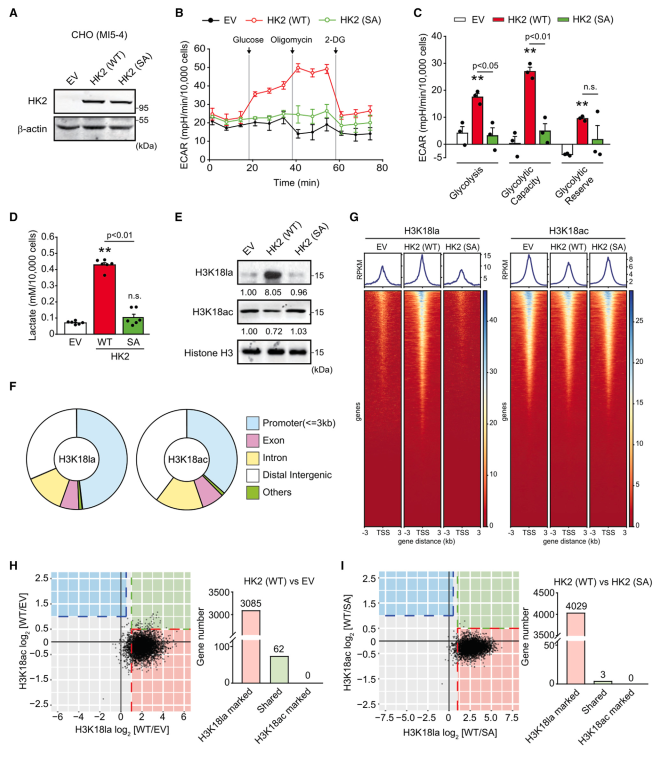
图1、在MI5-4 CHO细胞中,HK2表达诱导糖酵解、乳酸生成和组蛋白乳酸化
2、H3K18la标记基因参与基因表达调控和多种代谢过程
为了研究HK2表达和乳酸如何通过组蛋白乳酸化影响全局基因表达,作者首先通过外源性乳酸处理优化组蛋白乳酸化水平。作者发现,在EV细胞中,10 mM乳酸处理24小时,诱导的H3K18la水平与WT HK2细胞相当(图2A)。接下来,作者进行RNA测序(RNA-seq)来比较对照细胞(EV)、WT HK2细胞和乳酸处理的对照细胞(EV + Lac)的转录组。作者的差异表达基因(DEG)分析显示,外源性乳酸处理改变基因表达的模式与在WT HK2细胞中观察到的相似,通常上调1109个基因(WT HK2为68.5%,EV + Lac为62.7%),下调1532个基因(WT HK2为75.6%,EV + Lac为75.0%)(图2B, 2C)。接下来,作者确定了493个被HK2和乳酸处理上调的基因,这些基因在其启动子区域也被H3K18la标记(图2D)。在作者对基因本体(GO)生物过程(GOBP)术语的分析中,H3K18la标记的基因参与了基因表达、RNA和蛋白质代谢过程以及蛋白质修饰过程的调控(图2E)。通过qPCR证实,在EV、WT HK2、SA HK2和EV + Lac细胞中,选择了富含GOBP项的基因表达,包括冠层FGF信号调节因子2 (Cnpy2)、转录延伸因子Supt4a、富含丝氨酸和精氨酸的剪接因子11 (Srsf11)、线粒体转录拯救因子1 (Mtres1)、金属蛋白酶组织抑制剂1 (Timp1)和富含小脯氨酸蛋白1a (Sprr1a)。此外,在所选基因的启动子处验证了H3K18la和H3K18ac峰,在WT HK2细胞中观察到H3K18la峰明显增加,H3K18ac峰略有下降(图2G)。为了进一步证明HK2表达与组蛋白乳酸化之间的关系,作者生成了表达诱导HK2的MI5-4细胞。根据ECAR和耗氧率(OCR)的升高,证实了诱导HK2表达引起的糖酵解活性。与作者关于HK2组成表达的研究结果一致,作者观察到诱导可诱导的HK2表达也诱导了H3K18la,同时诱导了基因表达,而将HK2表达降低到基础水平则消除了这种作用。综上所述,这些结果表明HK2表达可提高糖酵解活性和乳酸生成,从而通过H3K18la影响基因表达。
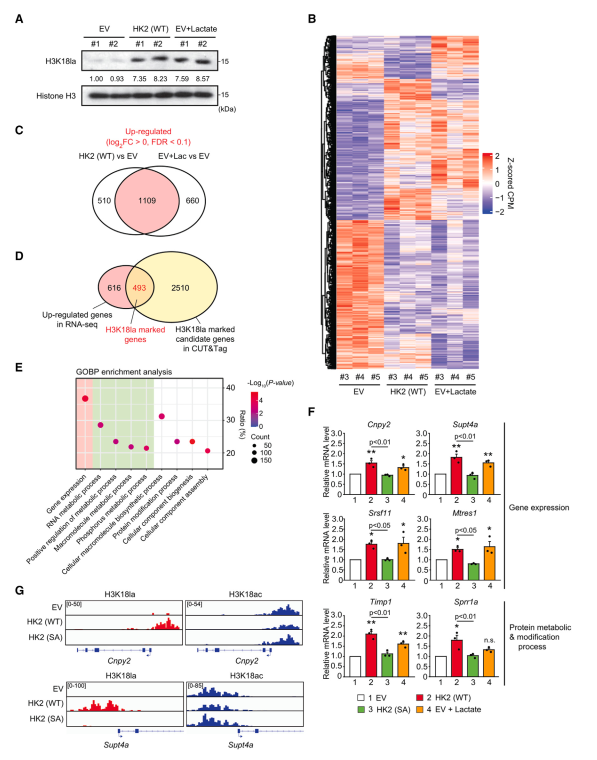
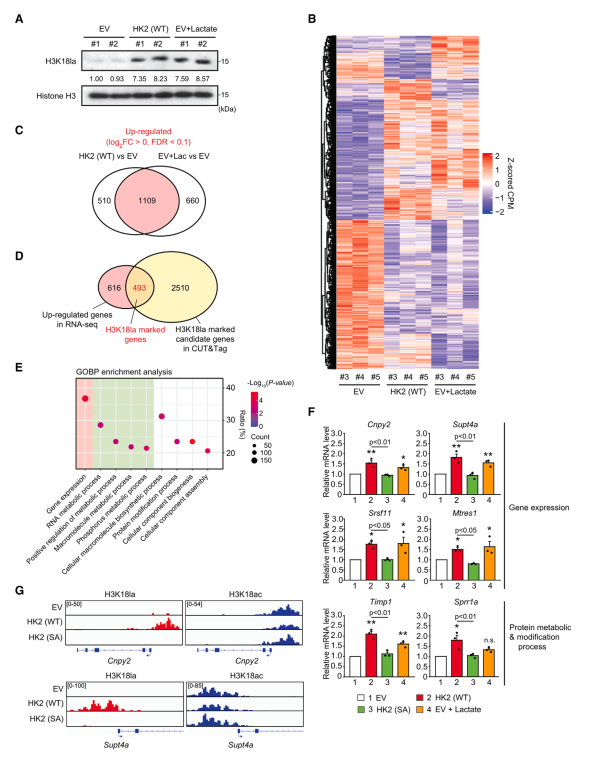
图2、HK2/ h3k18la特异性基因参与MI5-4 CHO细胞的基因表达和多种代谢过程的调控
3、H3K18la在活化的小鼠和HSC中被诱导
最近的研究表明,在慢性炎症和纤维化过程中,糖酵解的增强不仅局限于癌细胞,也发生在肝、肺和肾脏的成纤维细胞和肌成纤维细胞中。肝损伤后,静止的HSC被激活,成为纤维化肝脏中肌成纤维细胞的主要群体。尽管大量研究表明,通过诱导各种糖酵解酶,HSC的激活伴随着乳酸的产生,但乳酸是如何与HSC的激活联系在一起的仍是未知的。基于上述在MI5-4 CHO细胞中的发现,作者假设在HSC激活过程中,产生HK2的乳酸可能通过组蛋白乳酸化影响基因表达。为了验证这一假设,作者首先在CCl4注射或胆管结扎(BDL)引起的小鼠肝纤维化模型中证实了HK2的表达。作者观察到,在纤维化肝脏中,HK2的表达主要与表达a- SMA的肌成纤维细胞共定位。随后,作者用其他HSC激活诱导基因(Col1a1和Timp1)评估了HK2的mRNA水平,并观察到纤维化肝脏中HK2与HSC激活诱导的基因表达水平呈正相关。接下来,作者从小鼠肝脏中分离原代HSC。作者的免疫印迹分析显示,在注射ccl4的小鼠肝脏的原代HSC中,HK2的表达被强烈诱导,而在健康肝脏的HSC中已经表达的HK1,仅在活化的HSC中被轻微诱导(图3A)。同样,在静止的HSC中检测不到HK2的表达,但在培养3天和7天的小鼠原代HSC中,作者观察到HK2的强烈表达和a-SMA的表达(图3B)。在ccl4注射小鼠肝脏的原代HSC、BDL小鼠肝脏的HSC(公开可访问的RNA-seq数据集GEO: GSE154170)和体外培养7天的HSC(公开可访问的微阵列数据集GEO: GSE34949)中,也验证了HK2而非HK1的强诱导作用。然而,肝脏中主要的HK亚型,葡萄糖激酶(GCK),在小鼠原代肝细胞中表达,而在静止和激活的HSC中不表达(图S3K)。接下来,对ECAR、乳酸生成和细胞乳酸水平的测量显示,活化的HSC中糖酵解活性明显升高(图3C),导致体内和体外活化过程中H3K18la诱导(图3D和3E)。重要的是,除了H3K18la外,H3K9la、H3K14la、H4K8la和H4K12la在体外激活的HSC中也被诱导表达。与H3K18la不同,H3K18ac存在于静止的HSC中,其水平在体内激活的HSC中保持不变,但在体外激活时被抑制,这表明HSC激活可能需要H3K18la而不需要H3K18ac。与组蛋白乳酸化和乙酰化一致,作者还发现非组蛋白乳酸化主要在体外活化的HSC中升高,但乙酰化主要降低。为了在人类临床环境中加强作者的发现,作者对肝硬化患者的人肝组织进行了双重免疫荧光染色。患者肝脏免疫染色显示,82.9%±3.1%的a-SMA+细胞表达HK2, 63.0%±5.2%的细胞表达H3K18la, 78.2%±3.6%的COL1a1+细胞表达HK2, 66.6%±5.1%的细胞表达H3K18la(图3F),支持HSC活化与HK2表达和H3K18la存在的联系。总之,这些结果表明,诱导的HK2表达和糖酵解活性可能通过H3K18la在小鼠和人活化的HSC中发挥作用。
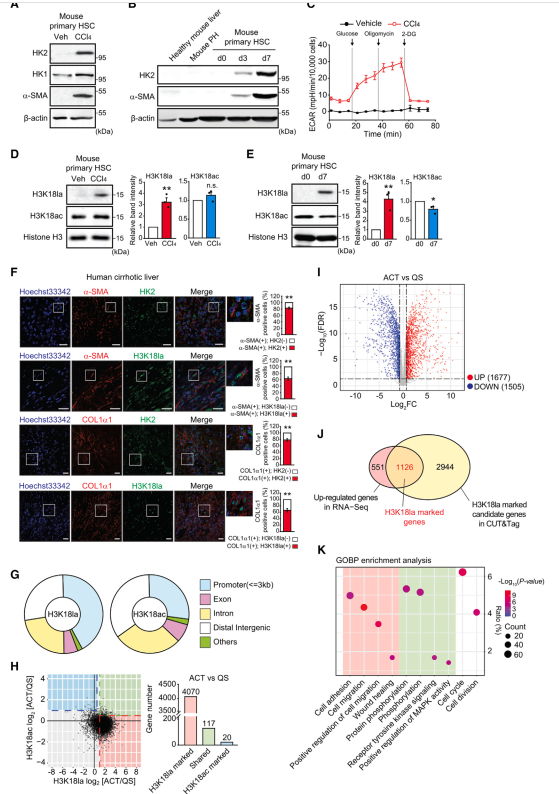
图3、在HSC活化过程中,HK2/H3K18la被诱导并调控基因表达
4、H3K18la富集于活化HSC中诱导的基因的启动子区域
为了进一步确定H3K18la在HSC激活过程中是否在基因表达中发挥作用,作者对刚从健康肝脏分离或培养6天的小鼠原代HSC进行了全基因组的CUT&Tag。有趣的是,活化的HSC中的H3K18la主要分布在启动子区域(42.5%),而H3K18ac在启动子区域的分布程度要小得多(27.4%),甚至低于包含子(28.2%)和远端基因间区域(34.5%)(图3G)。这些发现表明,H3K18la,而不是H3K18ac,在HSC激活过程中,在基因表达的诱导中起主要作用。通过分析启动子区域,作者确定了4070个基因为H3K18la标记的候选基因,117个基因共享H3K18la和H3K18ac,只有20个基因为H3K18ac标记的候选基因(图3H)。因此,激活的HSC中的绝大多数基因在启动子区域被H3K18la标记。为了鉴定活化HSC中H3K18la标记的基因,作者对培养6天的原代HSC进行了RNA-seq,结果显示,在活化过程中,1677个基因显著上调,1505个基因显著下调(图3I)。最后,作者鉴定出1126个基因在启动子区域被H3K18la诱导而上调(图3J)。GOBP term分析显示,这些H3K18la标记的基因参与了细胞粘附、迁移和伤口愈合(图3K),提示H3K18la可能调控代表HSC主要特征的关键基因。
5、HK2基因缺失通过降低H3K18la来抑制HSC的激活,而H3K18la可以通过外源性乳酸而不是醋酸盐的补充来恢复
鉴于作者的研究结果,激活的HSC通过诱导糖酵解以及HK2表达和H3K18la进行代谢重编程,作者推测HK2消融通过抑制H3K18la来阻碍HSC的激活。为了验证作者的假设,作者从HK2f/f小鼠的肝脏中分离出原代HSC,并通过暴露于表达Cre重组酶与eGFP融合的腺病毒(Ad-GFP-Cre)进行基因缺失,然后培养6天。HSC中HK2缺失使ECAR、乳酸生成和细胞乳酸水平降低约60%(图4A-4C)。OCR也显著降低,表明HK2的表达促进了HSC激活时糖酵解的主要部分,以提供丙酮酸,丙酮酸在细胞呼吸发生的线粒体中被还原为乳酸并氧化。此外,这种由HK2缺失引起的代谢扰动抑制了a-SMA的表达和增殖,但没有引起细胞死亡(图4D)。正如预期的那样,腺病毒Cre转导不影响WT原代HSC的激活(图4D)。在确定了HK2消融对糖酵解活性和HSC激活的影响后,作者接下来检测了H3K18la和H3K18ac的水平。有趣的是,HK2缺失细胞的H3K18la和H3K18ac水平低于活化的HSC,但外源性乳酸处理恢复了H3K18la,而H3K18ac进一步降低(图4E)。与此一致的是,CUT&Tag结果的热图分析显示,激活小鼠原代HSC中,H3K18la在基因的TSSs附近被显著诱导,通过HK2缺失而减弱,在添加乳酸后恢复(图4F)。相比之下,H3K18ac在活化的HSC中减少,并通过HK2缺失和乳酸处理进一步减少(图4F)。为了进一步研究H3K18la标记的基因是否依赖于HK2的表达以及乳酸处理,作者比较了活化的WT型HSC和活化的HK2敲除(KO)型HSC中H3K18la标记的基因的表达。作者发现,在活化的HK2-KO细胞中,下调基因增加了大约3倍(图4G)。乳酸处理逆转了大多数依赖于HK2表达的基因的表达(图4H)。GOBP term分析显示,这些依赖于HK2/H3K18la和乳酸处理的真实基因与细胞粘附、迁移、细胞外基质组织和TGF-b受体信号通路有关,表明HK2/H3K18la在调节HSC激活诱导基因中起关键作用。事实上,在活化的HSC中,H3K18la峰在关键诱导基因的启动子处高度富集,而在静止和HK2-KO细胞中则没有,但在HK2-KO细胞中,乳酸处理恢复了H3K18la峰(图4I、4J)。与作者在tss附近的全基因组H3K18ac峰的发现一致,在活化的HK2-KO细胞和乳酸处理的HK2-KO细胞中,H3K18ac峰在基因的启动子处逐渐被抑制。已知补充乙酸可以增强组蛋白乙酰化和染色质可及性。为了评估乳酸和醋酸盐处理是否能恢复HK2-KO HSC中的基因表达,作者测量了HSC激活诱导基因的转录水平。令人惊讶的是,乳酸处理上调了被HK2缺失抑制的基因表达,而醋酸处理没有(图4K)。同样,活化的HSC的侵袭能力因HK2缺乏而降低,但乳酸处理使其恢复,而醋酸处理未恢复表型(图4L)。总的来说,作者的研究结果表明,HK2缺失抑制HSC激活,乳酸处理(可能通过H3K18la)可以挽救HSC激活,而醋酸处理则不能。
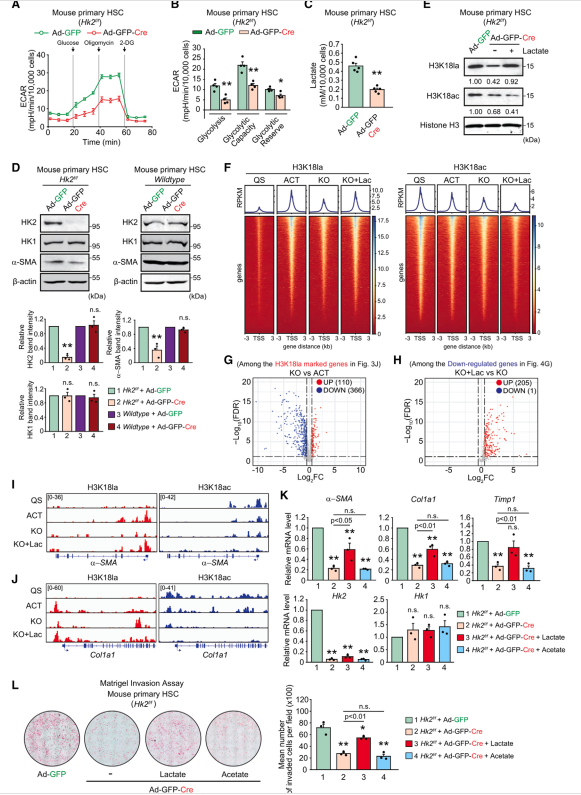
图4、HK2基因缺失通过降低H3K18la来抑制HSC的激活,而外源性乳酸处理可以挽救H3K18la,而醋酸处理则不能
6、抑制外源性乳酸产生可以抑制HSC的激活
为了继续研究组蛋白乳酸化与HSC激活之间的关系,作者进一步评估了乳酸生成药物抑制剂治疗后基因表达的激活。草酸酯通过抑制乳酸脱氢酶(LDH)抑制丙酮酸产生乳酸,而DCA通过抑制丙酮酸脱氢酶激酶(PDHK)增加丙酮酸转化为乙酰辅酶a而不是乳酸来抑制乳酸的产生(图5A)。正如之前报道的那样,草酸酯和DCA处理显著降低了乳酸生成和细胞乳酸水平(图5B、5C),这种干预抑制了小鼠原代HSC的细胞增殖,而不引起细胞死亡(图5D)。草酸酯和DCA处理有效地降低了H3K18la和HSC激活诱导基因的表达,乳酸可以挽救HSC激活诱导基因,而乙酸补充不能(图5E-5H)。为了进一步确定LDH与HK2诱导的组蛋白乳酸化和随后的基因表达之间的直接关系,作者在小鼠原代HSC中转染了靶向LDH的小干扰RNA (siRNA)。与LDH抑制剂处理一致,Ldha的siRNA敲低降低了细胞乳酸水平和H3K18la以及HSC激活诱导的基因表达,乳酸处理恢复了细胞乳酸水平,而醋酸处理没有恢复。TGF-b1被认为是在HSC纤维化信号通路中起重要作用的关键细胞因子。最近,越来越多的证据表明TGF-b1也通过增强HSC和成纤维细胞的糖酵解通量参与代谢重编程。因此,作者研究了TGF-b1对Lx-2细胞活化的影响。正如预期的那样,TGF-b1增加了乳酸的产生,这一作用被草酸盐或DCA处理所减弱。将葡萄糖水平从25 mM降低到5 mM会降低H3K18la,但TGF-b1在两种葡萄糖浓度下均会增加H3K18la,同时降低H3K18ac。正如预期的那样,TGF-b1诱导a-SMA基因表达和COL1A1基因表达。作者随后的时间过程研究发现,在TGF-b1处理后的较晚时间点(6-24 h), H3K18la持续升高,这与HSC激活诱导基因的显著上调相关。相比之下,H3K18ac水平在1 h时略有升高,但在TGF-b1处理后,这种升高随后持续降低。为了确定TGF-b1对Lx-2细胞中a-SMA基因启动子组蛋白修饰的影响,作者采用ChIPqPCR分析了启动子区域组蛋白的乳酸化和乙酰化。与作者的免疫印迹结果一致,TGF-b1增加了a-SMA启动子上的H3K18la,但降低了H3K18ac。接下来,作者发现草酸酯和DCA降低了H3K18la,抑制了TGF-b1升高的a-SMA表达。作者进一步证实这种抑制不是由于细胞死亡。在作者的拯救实验中,乳酸可以恢复激活诱导基因的表达减少,而醋酸不能恢复。作者的研究结果表明,干扰乳酸的产生会降低H3K18la和HSC激活诱导基因的表达,乳酸可以恢复,而乙酸不能恢复。综上所述,这些结果表明组蛋白乳酸化在HSC激活和随后的基因表达诱导中起着关键作用。
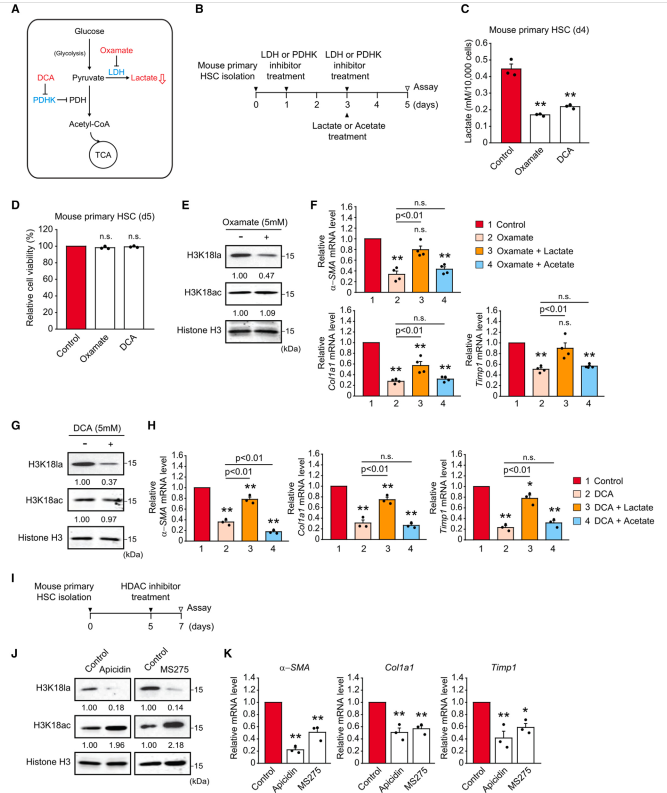
图5、通过抑制乳酸生成或I类HDAC抑制剂治疗降低H3K18la,使造血干细胞失活
7、一类HDAC抑制剂可提高H3K18ac,抑制H3K18la,从而抑制HSC激活诱导基因的表达
组蛋白去乙酰化酶(hdac)催化从组蛋白赖氨酸残基中去除乙酰基,导致染色质凝聚,抑制基因转录。先前的研究表明,在体外和体内,抑制仅局限于细胞核的I类hdac可使HSC失活。考虑到作者的研究结果,H3K18la是一种新的组蛋白修饰,反映了HSC激活诱导基因的表达,作者假设I类HDAC抑制剂可能诱导H3K18ac, H3K18la与H3K18la在组蛋白H3上相同的赖氨酸18残基上竞争。首先,作者用I类HDAC抑制剂apicidin和MS275 (entinostat)处理活化的小鼠原代HSC(图5I)。抑制剂处理增加了H3K18ac,但降低了H3K18la,表明赖氨酸18的乙酰化和乳酸化之间存在竞争(图5J)。其次,apicidin和MS275处理显著降低了基因表达和增殖的激活,这些影响不是由于细胞死亡(图5K)。为了进一步探索组蛋白乙酰化和乳酸化之间竞争的可能性,作者用乳酸和乙酸处理Lx-2细胞。单独乳酸处理增加了a-SMA的表达,而单独醋酸处理显示相反的效果。有趣的是,当添加醋酸盐时,乳酸诱导的a-SMA表达被消除。事实上,H3K18la在乳酸处理下被高度升高,但当添加醋酸盐时,这种升高被抑制,因此H3K18ac水平恢复。因此,作者的研究结果表明,I类HDAC抑制剂可能通过上调H3K18ac而失活HSC, H3K18la与H3K18la竞争,导致H3K18la依赖性基因表达下调。
8、HSC特异性和系统性的HK2缺失在体内抑制肝纤维化
为了进一步证实HK2/H3K18la在肝纤维化背景下在HSC中的作用,作者首先生成HK2f/f;LratCre小鼠,其中HK2可以在HSC中特异性删除。小鼠注射CCl4 3周,诱导肝纤维化(图6A)。Sirius red和Masson’s三色染色显示,当HSC中缺失HK2时,肝脏中CCl4引起的胶原积累明显减少(图6B和6E)。在胶原沉积的同时,a-SMA在HSC特异性HK2-KO小鼠肝脏中的表达显著降低(图6B和6D)。为了验证H3K18la水平是否反映了体内HSC的激活,作者在给药CCl4后从HSC特异性HK2-KO小鼠的肝脏中分离小鼠原代HSC。作者证实,CCl4给药后小鼠肝脏原代HSC中诱导的H3K18la水平因缺乏HK2而显著降低(图6C)。然而,H3K18ac水平没有改变。接下来,ALT(丙氨酸转氨酶)和AST(天冬氨酸转氨酶)的测定显示,HSC中HK2的缺失对肝毒性没有显著影响(图6F)。考虑到外源性乳酸补充在体外挽救了HK2缺失的HSC的HSC激活表型,作者接下来通过注射2 g/kg乳酸(腹腔注射)来评估乳酸对HSC特异性HK2-KO小鼠的影响,连续注射CCl4 3周,每隔一天一次。有趣的是,作者的Sirius red和a-SMA免疫染色结果显示,体内添加乳酸后,HSC特异性HK2-KO小鼠肝脏中胶原沉积减少和a-SMA表达恢复。总之,作者的研究结果表明,HSC特异性缺失HK2可减轻HSC中的H3K18la并抑制肝纤维化,而体内慢性和全身乳酸治疗可恢复胶原沉积和a-SMA表达。作者之前的研究表明,成年小鼠的全身HK2缺失对乳腺癌和肺癌具有良好的耐受性和治疗性,没有不良的生理后果。接下来,作者用CCl4治疗系统性HK2缺失小鼠3周(图7A)。值得注意的是,作者的Sirius red和Masson’s三色染色结果显示,在系统性缺失HK2后,肝脏中的胶原积累明显受到抑制(图7B和7D)。此外,证实a- SMA表达显著减少(图7B和7C)。为了进一步分析系统性HK2缺失的抗纤维化作用,作者接下来在注射他莫昔芬后建立了bdl诱导的HK2f/f;UBCCreERT2小鼠纤维化模型(图7F)。与作者之前的体内实验结果一致,系统性缺失HK2有效抑制了bdl小鼠肝脏中胶原的积累和a-SMA的表达(图7G-7I)。在ccl4诱导和bdl诱导的纤维化模型中,血清ALT和AST活性在HK2熟练组和HK2缺乏组之间相似,这表明体内系统性的HK2缺失不会引起显著的细胞毒性(图7E和7J)。接下来,为了进一步了解系统性HK2缺失对治疗潜力的影响,作者首先用CCl4治疗3周诱导肝纤维化,然后尝试系统性地删除HK2(图7K)。在系统性缺失HK2的小鼠肝脏中,Sirius红和Masson三色染色显示胶原沉积明显减少(图7L和7M)。作者观察到,在肝纤维化建立后,当全身缺失HK2时,a-SMA的表达也减少(图7N)。 此外,在诱导肝纤维化后,删除HK2对血清ALT和AST活性没有明显影响(图7O)。综上所述,作者的研究结果表明,靶向HK2可以系统性地抑制HSC的激活;因此,HK2可能是肝纤维化的治疗靶点。
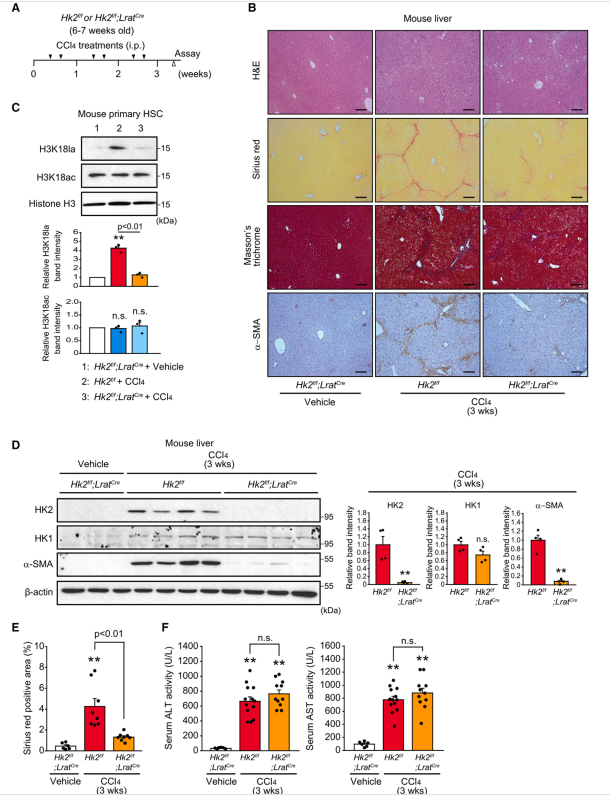
图6、造血干细胞中特异性缺失HK2可改善ccl4诱导的肝纤维化
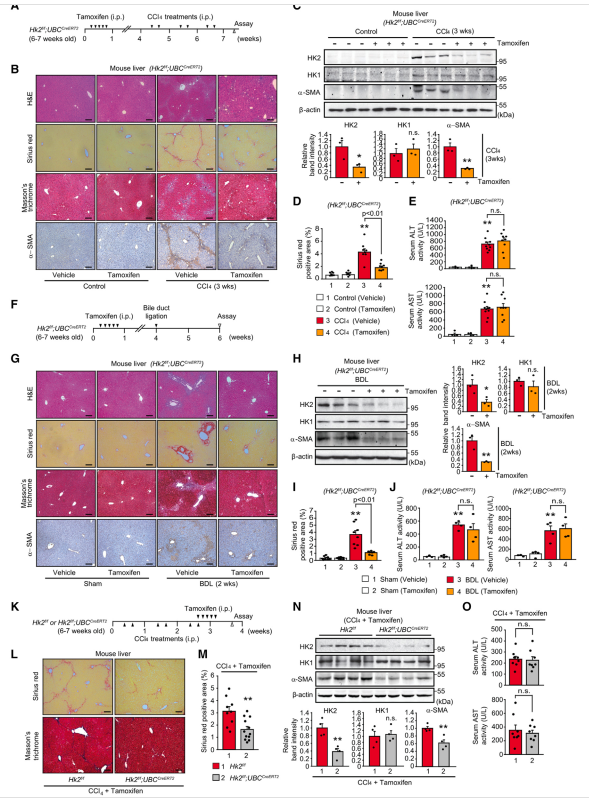
图7、体内系统缺失HK2对ccl4和bdl诱导的肝纤维化的抑制作用
结论
总之,作者发现HK2介导的乳酸生成通过组蛋白乳酸化影响基因表达。作者发现在活化的HSC中诱导HK2的表达对于HSC的激活至关重要,并且由HK2表达引起的组蛋白乳酸化决定了HSC激活后的基因表达。此外,作者的研究结果表明,干预HK2/H3K18la轴是肝纤维化的一种潜在治疗策略。
实验方法
WB, qPCR,IHC,IF,RNA-seq,CUT&Tag,CHIP-qPCR,CHIP-seq,ECAR,OCR,代谢组学
参考文献
Rho H, Terry AR, Chronis C, Hay N. Hexokinase 2-mediated gene expression via histone lactylation is required for hepatic stellate cell activation and liver fibrosis. Cell Metab. 2023 Aug 8;35(8):1406-1423.e8. doi: 10.1016/j.cmet.2023.06.013. Epub 2023 Jul 17. PMID: 37463576.