






MAF扩增通过表观遗传重塑许可ERα驱动乳腺癌转移
MAF扩增增加乳腺癌(BCa)转移的风险,其机制尚不清楚,但具有重要的临床意义。雌激素受体阳性(ER+) BCa的生长和转移都需要雌激素,尽管其机制尚不清楚。在这里,作者整合了蛋白质组学、转录组学、表观基因组学、染色质可及性和人类和同基因小鼠BCa模型的功能分析,表明MAF直接与雌激素受体α (ERα)相互作用,从而促进了一种独特的染色质景观,有利于转移扩散。作者确定了在雌激素暴露后以MAF依赖方式重新许可的促进转移的基因。组蛋白去甲基化酶KDM1A是表观基因组重塑的关键,它促进了促转移性MAF/雌激素驱动基因表达程序的表达,而KDM1A活性的丧失阻止了这种转移。因此,作者已经确定MAF/雌激素介导的转移的分子基础需要来自全身环境的遗传、表观遗传和激素信号,这些信号影响BCa细胞转移的能力。本文于2023年11月发表于《Nature Cell Biology》, IF: 21.3,Q1。
技术路线:

主要实验结果:
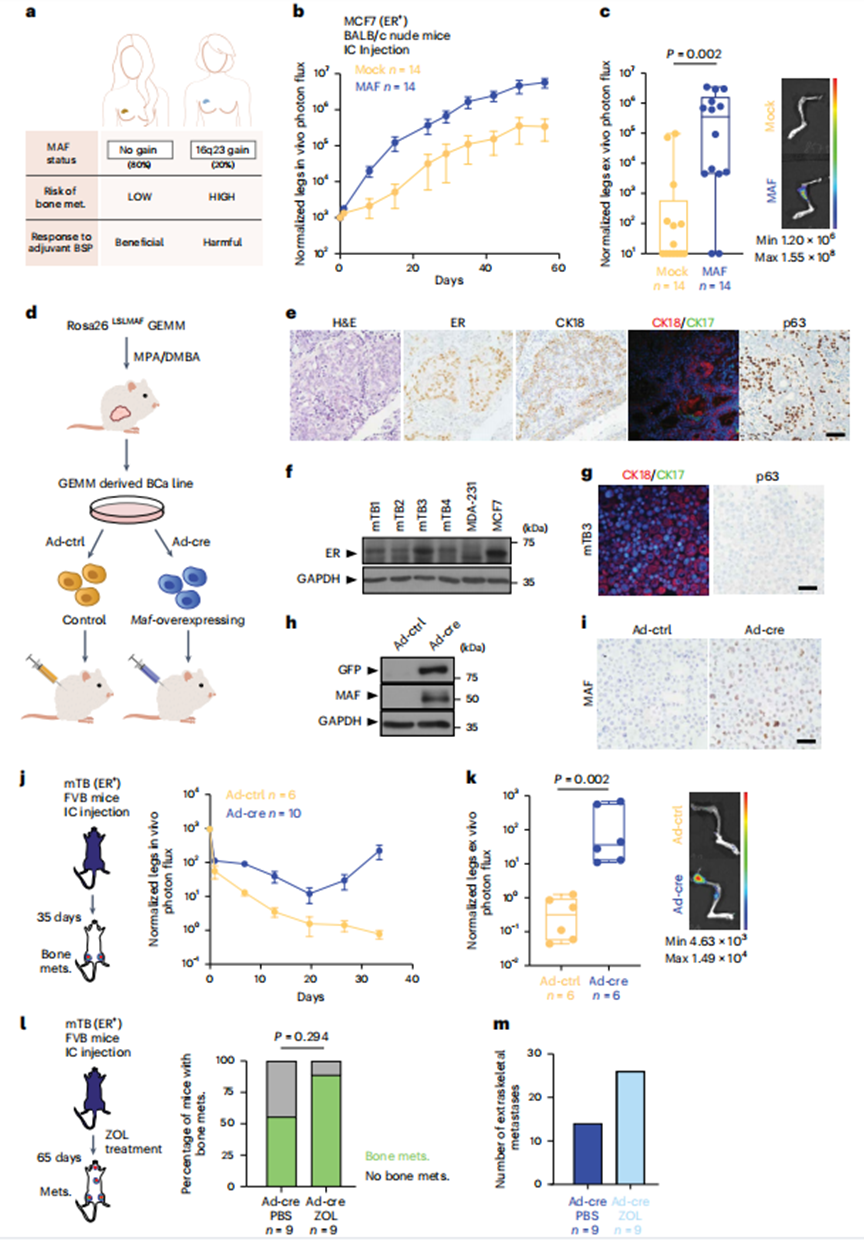
1、MAF过表达促进ER+BCa细胞骨转移
为了分析MAF在ER+BCa中的作用,作者首先分析了ER+BoM2 BCa细胞,这些细胞是从亲本ER+MCF7 BCa细胞群中选择的小鼠骨趋向性细胞,因为它们在体内驱动转移的能力。值得注意的是,MCF7细胞可能有局限性,但与其他腔内BCa细胞(如T47D和ZR-75)相比,不存在内质网和雄激素受体活性之间的混淆相互作用。在体内,MAF过表达足以显著增加MCF7细胞接种于胸腺裸鼠左心室时的骨转移率,而在其他部位无转移差异(图1b、c)。体内肿瘤生长需要补充雌激素。接下来,作者测试了MAF过表达是否以E2依赖的方式增强体外细胞增殖。与模拟E2处理的细胞或激素剥夺(HD)对照相比,MAF阳性细胞中E2刺激的细胞增殖增强,表明MAF表达与E2之间存在生物学相互作用。
接下来,作者建立了一个条件Maf过表达敲入小鼠模型(Rosa26LSLMAF),以:(1)提供一种独立的方法,(2)排除人BCa细胞系中获得性遗传改变的存在,(3)直接测试MAF表达增加对E2依赖性BCa转移的影响。为了诱导小鼠BCa肿瘤,作者用醋酸甲孕酮和7,12-二甲基苯并蒽(MPA-DMBA)处理Rosa26LSLMAF小鼠,使用已建立的BCa化学致癌方案(图1d,e)。作者在体外扩增小鼠肿瘤源性细胞系(mTB细胞);值得注意的是,这些保留了内质网和角蛋白的表达(图1f,g)。然后用Ad-Cre颗粒感染诱导Maf(或不诱导,作为对照),作者得到了具有相同基因组背景的等基因细胞系对,无论是否有Maf过表达(图1d,h,i)。无论注射部位如何,Maf阳性的mTB细胞比Maf阴性的对照mTB细胞产生更多的骨转移(图1j,k)。最后,作者证实Maf诱导的骨转移对双磷酸盐治疗是不耐受的,并且骨外转移(肺、肝、肾、卵巢和脑)显著促进,如患者所述(图1l,m)。
总的来说,这些结果证实了MAF -阳性细胞在骨定植过程中具有竞争优势,可以被MAF -阴性细胞所吸收,产生以MAF -阳性克隆为主的异质性转移。因此,作者接下来重点阐明(1)E2信号与ER+BCa细胞中MAF -过表达之间相互作用的分子机制,以及(2)这些相互作用如何促进骨转移。
 MAF扩增驱动E2/ER信号依赖性BCa转移
MAF扩增驱动E2/ER信号依赖性BCa转移
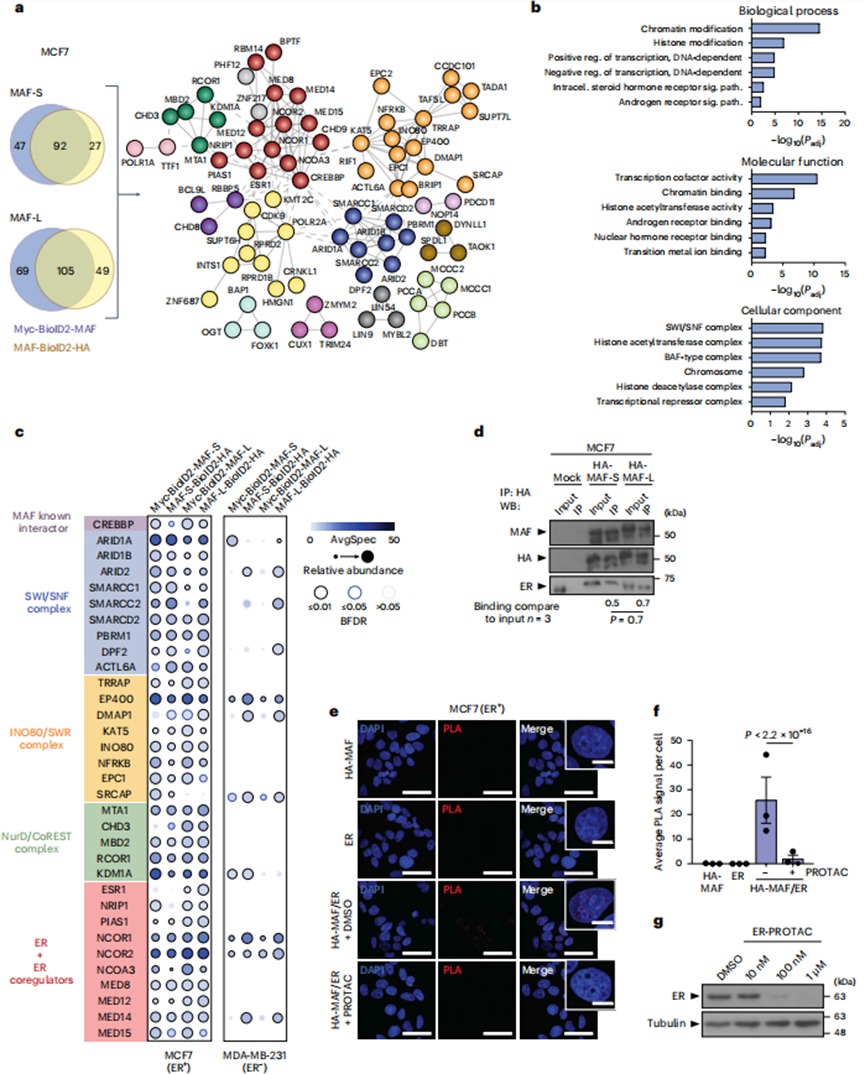
2、MAF、ER和染色质因子在ER+BCa细胞中相互作用
MAF编码AP1家族转录因子(TF),其DNA结合(因此其促转移活性)可能取决于与细胞环境特异性表达的相互作用伙伴。为了以无偏倚的方式鉴定活BCa细胞中与MAF相互作用的蛋白质,作者进行了邻近依赖的生物素鉴定(BioID2),可以检测到微弱和短暂的相互作用。虽然在功能上没有区别,但作者同时使用了短(MAF-S)和长(MAF-L)亚型,以避免任何潜在的亚型依赖偏差。每个MAF异构体在框架内融合到BioID2的N或C端,分别产生MAF-BioID2- HA和myc-BioID2-MAF(图2a)。从含有BioID2- MAF或(作为对照)BioID2的生物素处理细胞中提取链霉亲和素后,通过串联质谱(nanoLC-MS/MS)鉴定共沉淀蛋白。作者获得了许多高置信度的交互作用因子(方法):139 N -末端标记的MAF-S,119 C -末端标记的MAF-S,174 N -末端标记的MAF-L,154 C -末端标记的MAF-L(图2a)。然后,作者选择了92个MAF-S条件共有的相互作用因子,以及105个MAF- L条件共有的相互作用因子,来定义一个126个MAF高置信度相互作用因子的网络(其中71个在所有四种条件下都是常见的)。这组126个基因中包括表征良好的MAF相互作用因子CREBBP,并且在已知蛋白质-蛋白质相互作用的高度互联网络中被强烈富集(P < 1 × 10−16),表明MAF相互作用因子之间存在生物相关复合物(图2b)。事实上,存在一些主要的染色质重塑复合物,如SWI/SNF、INO80、NurD和CoREST。GO富集分析还确定了影响基因表达的分子功能,包括激素受体信号家族的成员。值得注意的是,ER本身作为生物学相关的MAF相互作用物出现(图2a-c)。内质网和其他相互作用因子随后通过共免疫沉淀(co-IP;图2d)进行验证。为了确定ER的存在是否会影响MAF的相互作用,使用ER- MDA-MB-231 BCa细胞进行BioID(图2c)。在这些ER-细胞中,MAF保留了与SWI/SNF、INO80、NurD和CoREST染色质重塑复合物的某些组分的相互作用,但不与ER和/或关键ER共激活子相互作用(图2c)。总的来说,这些数据表明MAF和特定伙伴之间的相互作用依赖于细胞类型。
 MAF与内质网转录复合物相互作用
MAF与内质网转录复合物相互作用
3、MAF的N端转激活结构域与ER相互作用
接下来,作者使用HA标记的MAF- S和MAF- L在MCF7细胞中通过co-IP验证MAF的相互作用(图2d)。通过近距离连接试验(PLAs),作者证实了MAF-ER相互作用和共定位,与单一抗体对照相比,HA和ER抗体联合在细胞核中检测到和量化的荧光信号明显更高(图2e,f)。基于在MCF7 ER+BCa细胞中使用ER特异性PROTAC诱导ER降解,作者证实了MAF-ER相互作用的特异性(图2e-g)。然后,作者使用PLA通过比较全长MAF- L与缺少部分或全部转录激活域的截断版本(分别为ΔN-t 1, aa 85-403和ΔN-t 2, aa 120-403)来研究与ER相互作用的MAF的蛋白质结构域。然而,作者不能排除与ER AF1或AF2结构域的相互作用具有不同的作用,如前所述。
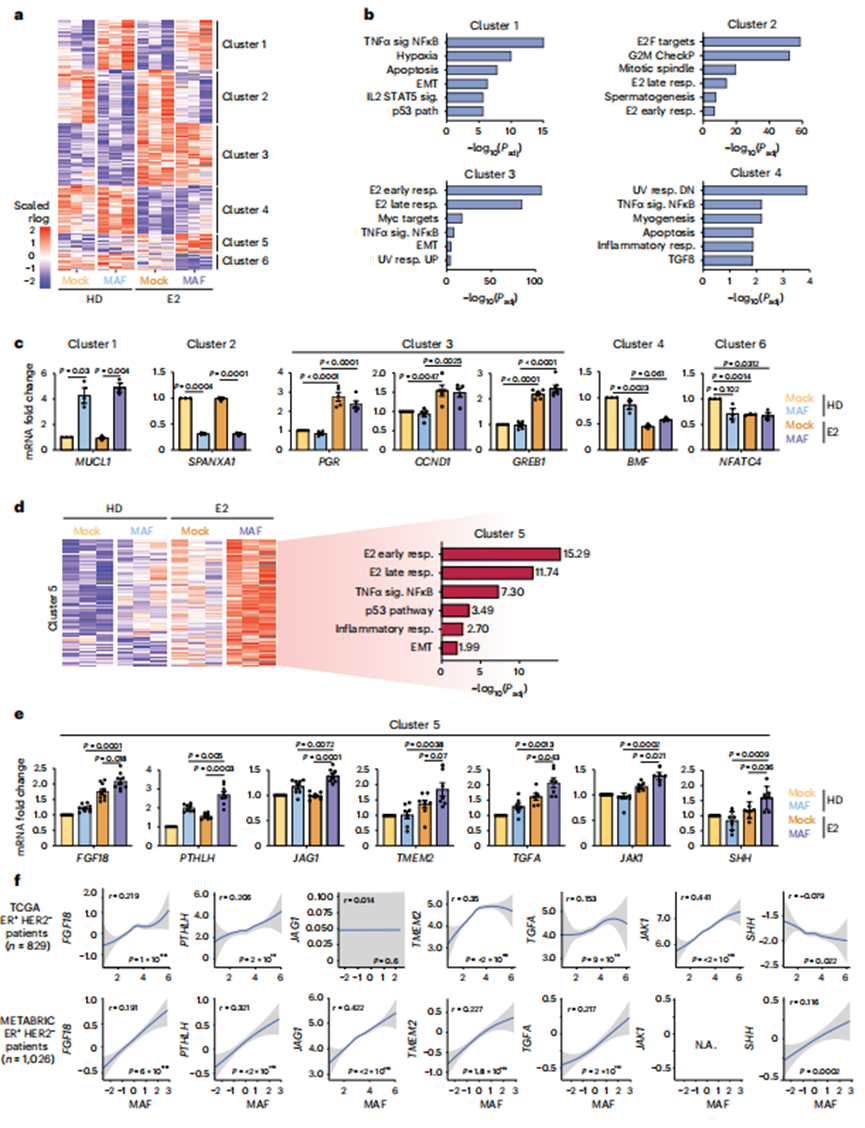
4、MAF-ER相互作用导致转录重编程
接下来,作者对激素剥夺(HD)或E2处理6小时的MCF7细胞进行RNA测序(RNA-seq)(图3a,b)。作者确定了转录变化需要(1)MAF过表达(簇1和2),(2)E2(簇3和4)和(3)MAF和E2(这里,MAF/E2依赖;簇5和6)(图3a)。与E2刺激的增殖一致,E2处理与E2早期和晚期基因应答(簇3)正相关(图3a,b),包括已建立的ER介导的应答基因GREB1, CCND1和PGR(图3c)。值得注意的是,MAF/E2依赖性基因标记(簇5)与E2早期和晚期反应、炎症和上皮向间质转化(EMT)基因反应呈正相关,包括PTHLH、JAG1、FGF18、TMEM2、TGFA、JAK1和SHH;这些基因产物支持转移适应能力,尤其是骨(图3d,e)。重要的是,作者观察到在ER+BCa患者基因表达数据集中,包括乳腺癌国际分子分类协会(METABRIC)和癌症基因组图谱(TCGA) BCa队列中,MAF的表达水平与FGF18、PTHLH、JAG1、TMEM2、TGFA、JAK1和SHH的表达水平呈正相关(图3f)。总的来说,这些数据表明MAF过表达在E2刺激下以ER依赖的方式扩大了ER+BCa细胞的转录库。作者提出MAF过表达支持BCa在增殖和原发肿瘤生长之外的进展,可能是通过直接激活骨微环境使其更容易接受转移状态。
 MAF调节E2/ ER诱导的转移转录基因程序
MAF调节E2/ ER诱导的转移转录基因程序
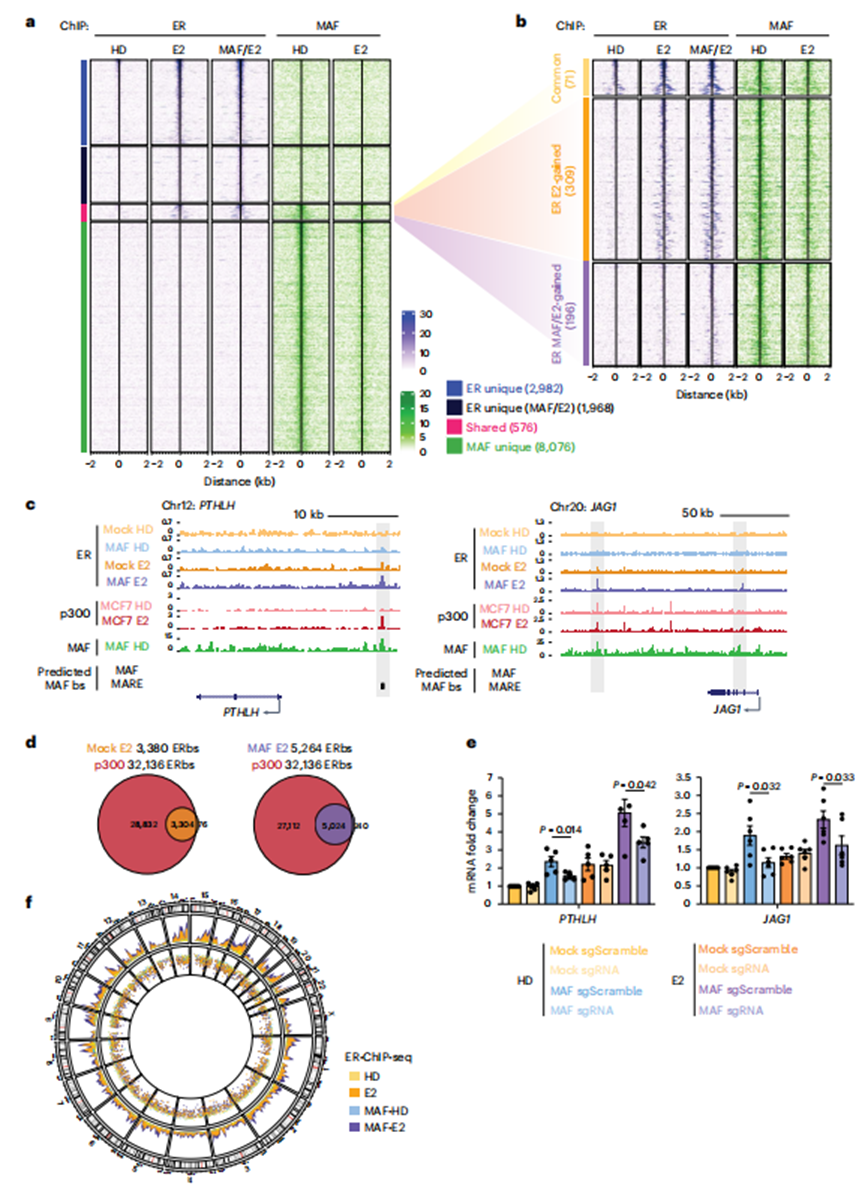
5、MAF在染色质上重新分配内质网以靶向转移相关基因
为了深入了解MAF的全基因组定位,并评估其在E2处理后与ERα的潜在直接相互作用,作者在对照和MAF过表达的MCF7 BCa细胞中进行了ERα和MAF染色质免疫沉淀和高通量测序(染色质免疫沉淀和测序,ChIP-seq)。事实上,只有在E2处理后,才观察到ER与染色质的广泛结合,这表明E2在内质网向染色质募集中的作用(图4a)。相反,MAF的结合在很大程度上独立于E2(图4a)。然而,有趣的是,在E2的作用下,ER和MAF都结合了一组共享的基因组位点(576),其中一些位点(196)仅在MAF过表达的细胞中被ER结合(图4a,b)。在基因组浏览器中对这些位点进行目视检查,发现E2诱导的MAF-E2依赖性ER与MAF-E2靶转移支持基因相关增强子中的未知靶点结合,如FGF18、PTHLH、JAG1、TMEM2、TGFA、JAK1和SHH(图4c;注意GREB1、CCND1和PGR被用作真正的ER/E2靶点)。这些假定的顺式调控元件在MAF共识结合基序(MARE和MAF)中富集,并且与增强子标记p300的ChIP-seq峰相吻合(图4d)。
接下来,作者使用CRISPR干扰(CRISPRi)来抑制PTHLH和JAGGED1转录起始位点(TSSs)附近的这些假定的MAF应答元件,以确认它们的功能作用。在使用特定的单导RNA (sgRNA)对共享的MAF/ER峰位点和dCas9失活后,MAF/ER依赖的转录对PTHLH和JAGGED1的转录诱导显著减弱,与E2无关(图4e)。总的来说,作者的数据表明MAF的存在直接或间接地增加了ER与染色质的结合。此外,MAF直接与ERα相互作用,因此其在BCa中的过表达与ER顺反组相关联并扩大(图4f)。作者假设MAF过表达,通过其与染色质重塑物的直接相互作用(图2c),启动染色质引物,响应E2,促进ER基因调控,最终促进ER+BCa的转移过程。
 MAF染色质结合与内质网重叠并扩展其结合
MAF染色质结合与内质网重叠并扩展其结合
6、ER/MAF协同提供了一个特定的染色质景观
接下来,作者在E2刺激下生成了高度转移的MAF阳性MCF7 BCa细胞的染色质可及性图。与对照细胞相比,MAF过表达和E2刺激都在细胞染色质状态上留下了明显的印记,这是通过染色质可及性的变化来测量的;作者可以定义MAF依赖簇A和B(分别为97和80个峰),E2依赖簇C和D(分别为40和76个峰)以及MAF/E2依赖簇E和F(分别为621和797个峰)(图5a-c)。MAF/E2依赖性簇特有的广泛染色质重塑在ER耗尽时丢失,主要是在带注释的启动子/ TSSs(图5d)。重要的是,与其他簇相比,MAF依赖簇A和E的启动子/TSS区域的ATAC(转座酶可及染色质测定)峰的宽度显着增加(图5e)。值得注意的是,MAF依赖的ATAC峰与BCa细胞中的组蛋白标记重叠,并与启动子区域的转录激活(H3K27Ac, H3K4me3)相关(图5f)。
为了确定这些鉴定出的染色质可及性峰的变化是否反映了观察到的转录变化(图3a),作者整合了ATAC-seq和RNA-seq数据集。MAF过表达E2处理细胞的ATAC-seq数据显示,MAF和E2同时调控表达的基因显著富集(图5g;集群A至F如图5a所示)。
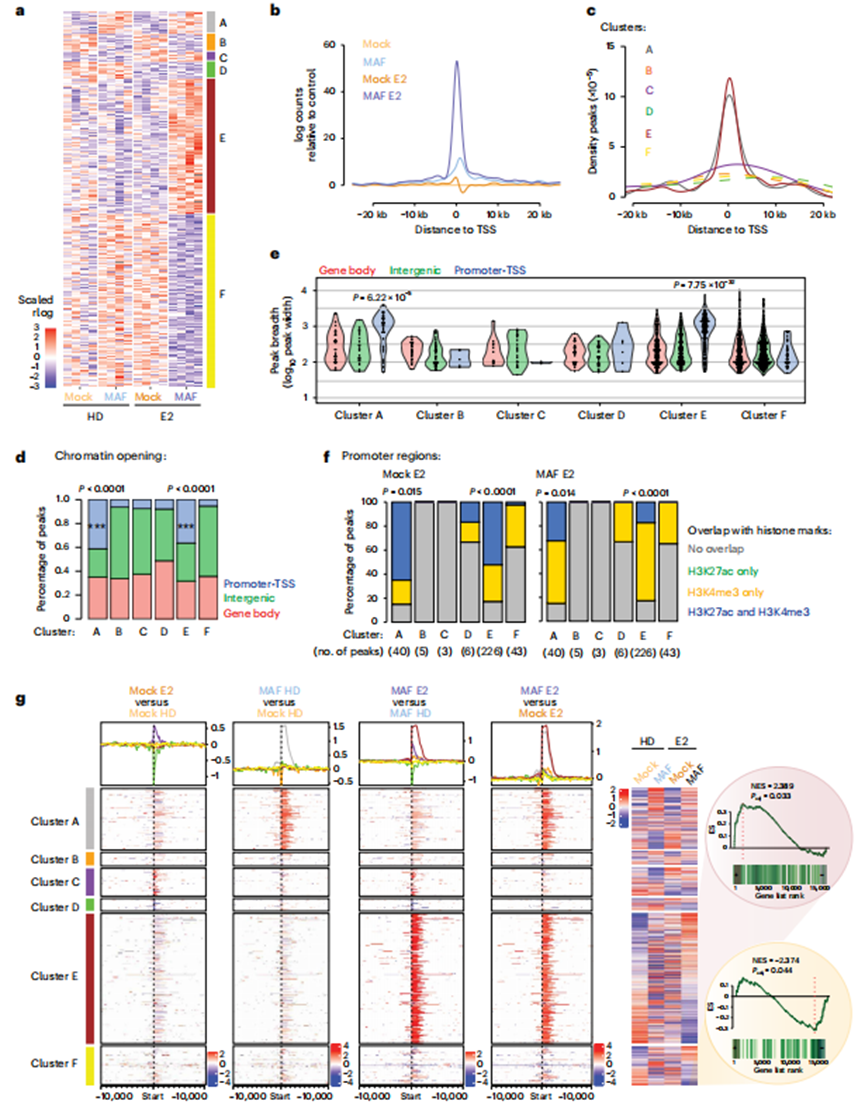 MAF扩增引起染色质景观的变化
MAF扩增引起染色质景观的变化
7、MAF和ER调控转移基因表达程序
为了将这些染色质结合事件与转录调控联系起来,作者使用先前描述的RNA-seq数据进一步表征了MAF/E2依赖性基因(图3a)。GO分析显示,E2上调的一些基因是已知的雌激素反应基因(如ple、MYC和细胞周期介质)。其他MAF/E2依赖基因属于以前未与E2信号相关的途径(图3b,d),包括与侵袭性和高度转移性BCa表型相关的基因(例如,EMT,炎症)。重要的是,MAF和ER之间共享的染色质结合位点是最近发现的E2靶向基因附近最富集的位点,以及经典的E2应答(图6a)。同样,在MAF/E2条件下,差异开放和关闭的染色质区域在ER ChIP峰中富集(开放峰363个,关闭峰193个)(图6b)。总的来说,这些数据表明,在本文发现的E2靶基因附近以前未被发现的顺式调控元件可能被MAF用于支持ERα结合并激活转录。
根据多个TCGA患者肿瘤中500 kb范围内RNA-seq和ATAC-seq信号的相关性,作者可以推断患者样本中MAF-ER结合直接控制的靶基因(而不仅仅依赖于基因接近性)。重要的是,作者发现,在MCF7细胞中同时被ER和MAF占据的患者乳腺肿瘤中,ATAC-seq峰通过启动子-增强子连接在功能上与E2靶基因表达的控制相关联,其程度高于偶然预期(例如,SOX9与近似随机排列相比,P < 0.0001;图6 c, d)。相比之下,单独由MAF结合的位点没有显示出这种实质性的富集(图6c)。总的来说,这些结果表明E2主要通过ER位点发出信号,然而,在MAF表达时,ER能够与MAF一起结合到以前未被发现的基因组位点,从而从经典的E2应答基因程序中扩展E2诱导的转录库。事实上,通过整合RNA-seq转录组学和MAF-和MAF/E2依赖性ER ChIP-seq数据产生的MAF-依赖性基因标记确定了ER+患者的一个子集,其最初经历骨骼复发的可能性更高(图6e)。
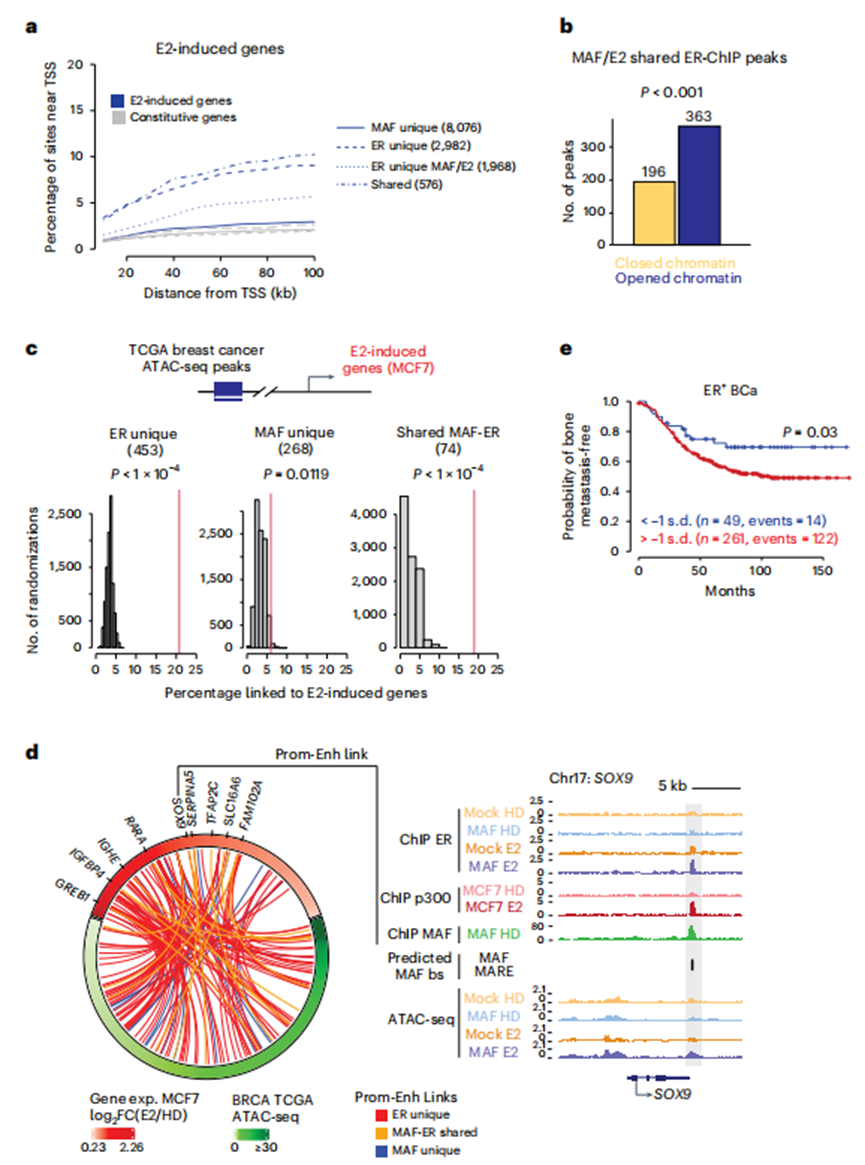 共享ER-MAF结合位点控制E2诱导的转移基因程序
共享ER-MAF结合位点控制E2诱导的转移基因程序
8、定义ER BCa转移的表观遗传开关
接下来,作者将重点放在高可信度的MAF相互作用物上,这些相互作用物可能通过组蛋白甲基化与MAF介导的全局染色质打开有关,并且以前与癌症有关。KDM1A在贝叶斯错误发现率(BFDR) < 0.001的相互作用组中进行了显著性分析(图2c),它编码一种黄素依赖的单胺氧化酶,可以去甲基化单和二甲基化赖氨酸(K),特别是组蛋白3和赖氨酸4和赖氨酸9 (H3K4和H3K9)。值得注意的是,高KDM1A活性存在于许多癌症类型中,包括BCa。为了确定在MAF过表达的情况下,抑制KDM1A活性对E2介导的转录重布线的影响,作者在MCF7 BCa细胞中进行了PLA测定和KDM1A和MAF的co-IP(图7a)。与对照细胞相比,KDM1A敲除的细胞PLA信号明显减少(具有短针乱置(shSc))(图7a-c)。这些结果证实了MAF直接与KDM1A结合,并表明这种相互作用有助于促进MAF - ER相互作用。
为了分析KDM1A敲低是否会影响MAF/ E2介导的基因应答,作者比较了HD与E2处理细胞的RNA-seq数据(图3a中簇5和6)(图7d)。在MAF过表达的细胞中,作者观察到缺乏KDM1A活性降低了E2/ ER诱导和MAF依赖性基因的表达(例如,FGF18, PTHLH, SOX9, TMEM2, JAK1和SHH)(图7d,e),但其他基因没有(图7d,e)。综上所述,这些结果表明KDM1A有助于MAF依赖性基因反应,包括MAF/ E2依赖性基因反应的一个子集。
 KDM1A抑制破坏E2/ER和MAF依赖的信号传导并阻止转移
KDM1A抑制破坏E2/ER和MAF依赖的信号传导并阻止转移
9、KDM1A阻断可拮抗MAF依赖性转移
接下来,作者测试了KDM1A的高选择性共价抑制剂iaddemstat (ORY -1001)是否可以阻断MAF介导的转移。经ORY-1001处理后,MCF7和mTB细胞显示H3K9me2积累,显示KDM1A去甲基化酶活性的功能性抑制(图7f,g)。为了测试KDM1A抑制是否会阻止表达MAF的BCa细胞形成骨转移,作者使用了体外骨培养阵列(BICA)。然后,作者将对照或表达Maf -的mTB细胞接种到免疫无能力的同基因小鼠(FVB背景)的胫骨中。值得注意的是,ORY -1001治疗显示骨病变的数量和大小均显著减少,但仅在表达Maf的组(图7i),这表明KDM1A介导的Maf表达细胞的表观遗传变化驱动了骨适应和转移。
结论:
作者报告了一种以前未知的MAF-ER相互作用,这种相互作用是由表观基因组机制驱动的,并有助于BCa临床相关的转移结果。这涉及MAF驱动的染色质扰动,大大扩展了ER转录范围,超出了目前描述的目标。总的来说,作者的研究结果支持E2存在下MAF扩增后ER+BCa骨转移增强。重要的是,作者证明了在ER+肿瘤中,MAF的扩增和过表达将典型细胞重新编程,使其包含以前未知的增强子和转录活性,从而促进骨转移。该程序重定向DNA可及性,并通过KDM1A介导的转录通道促进转移。这些数据表明,染色质失调是BCa转移的早期事件,而基因组扩增在这一过程中起着核心作用。作者的结果为探索KDM1A在这种临床背景下的抑制作用打开了大门。
实验方法:
Maf转基因小鼠的产生;Southern blot;长PCR分析;细胞培养;小鼠Bca诱导肿瘤及衍生细胞外植体;MAF过表达细胞的产生;慢病毒加工;邻近依赖生物素鉴定(BioID);网络分析;Dot-plot分析;BioID相互作用富集分析;免疫印迹法;免疫荧光;免疫共沉淀;聚乳酸实验;PROTAC用于目标降解;RNA-seq;预处理和差异表达分析;内质网降解的RNA-seq;RNA-seq(与敲除KDM1A后的样品比较);逆转录定量聚合酶链反应(qRT-PCR);患者队列的相关性和生存数据分析;体外BrdU掺入测定;组织病理学和免疫治化学;细胞增殖试验及半最大抑制浓度(IC50)测定;ATAC-seq;内质网降解的ATAC-seq;ATAC-seq和RNA-seq数据的整合;ER和MAF ChIP-seq;H3K27Ac和H3K4me3 ChIP-seq;ChIP-seq和RNA-seq数据的整合;超增强子的识别和分配;CRISPRi;循环肿瘤细胞;细胞竞争试验;BICA。
参考文献:
Llorente A, Blasco MT, Espuny I, Guiu M, Ballaré C, Blanco E, Caballé A, Bellmunt A, Salvador F, Morales A, Nuñez M, Loren G, Imbastari F, Fidalgo M, Figueras-Puig C, Gibler P, Graupera M, Monteiro F, Riera A, Holen I, Avgustinova A, Di Croce L, Gomis RR. MAF amplification licenses ERα through epigenetic remodelling to drive breast cancer metastasis. Nat Cell Biol. 2023 Nov 9. doi: 10.1038/s41556-023-01281-y. Epub ahead of print. PMID: 37945904.