






ADAM12调控的巨噬细胞胞葬促进抗肿瘤免疫
本研究中,作者在黑色素瘤、胰腺癌症和癌症小鼠模型的肿瘤边缘鉴定到诱导的慢循环ADAM12+ PDGFRα+间充质基质细胞(MSC)。使用诱导型谱系追踪和转录组学,证明代谢改变的ADAM12+ MSC通过过表达Gas6、Lgals3和Csf1等基因促进巨噬细胞胞葬和极化,从而诱导病理性血管生成和免疫抑制。ADAM12+细胞的基因缺失恢复了功能性肿瘤血管,减少了缺氧和酸中毒,使CAF正常化,诱导效应T细胞浸润,并抑制黑色素瘤和胰腺神经内分泌癌症的生长,这一过程依赖于TGF-β。在人类癌症中,ADAM12对具有高水平缺氧和先天耐药机制的患者以及与不良预后和耐药性相关的因素(如AXL)进行了分层。总之,这些数据表明,通过ADAM12耗竭肿瘤诱导的慢循环PDGFRa+ MSC可以恢复抗肿瘤免疫。该研究于2023年10月发表在《nature immunology》,IF 30.5。
技术路线:
主要研究内容:
1 ADAM12+ MSCs基因缺失恢复肿瘤免疫
为了在肿瘤发生过程中观察到ADAM12+细胞,作者用B16-OVA黑色素瘤细胞(MO5)皮下接种ADAM12-GFP小鼠。观察到ADAM12+ MSC(表达GFP)特异性定位于肿瘤边缘,这是一个富含基质细胞的过渡区,表达不同水平的PDPN和平滑肌肌动蛋白α2(αSMA+)、胶原细胞外基质(ECM)、T细胞、CD206+ 肿瘤相关巨噬细胞(TAM)和血管(图1a、b)。ADAM12在正常皮肤或CD45+肿瘤免疫细胞、CD31+内皮细胞或PDPN– PDGFRα–基质细胞中未检测到,但其在肿瘤周围血管附近的2-8%的PDGFRα+ PDPN+细胞中被诱导表达。ADAM12+细胞频率和绝对数量在肿瘤晚期降低(图1b、c)。ADAM12+ MSCs是PDGFRβ+、αSMA-和NG2lo或NG2-(一种周细胞标记物),且定位于ColiV+血管基底膜(BM)外,与周细胞形成对比。为了耗尽ADAM12+ MSC,作者用MO5黑色素瘤细胞接种ADAM12- DTR小鼠;在这些小鼠中,白喉毒素受体(DTR)在Adam12启动子的控制下表达。从肿瘤植入后10天开始,当肿瘤可触及时,ADAM12+细胞耗竭导致50%的肿瘤生长抑制(图1d)。相反,在肿瘤发生的初始阶段,ADAM12+细胞的耗竭并不能抑制肿瘤生长(图1e),这与基质细胞的初始饲养作用相反。从第10天起,缺乏ADAM12+细胞的肿瘤干扰素-γ(IFN-γ)产生CD8+ T细胞和自然杀伤(NK)细胞浸润增加(图1f),而CD4+ T细胞、调节性T细胞(Treg细胞)、嗜酸性粒细胞、中性粒细胞、树突状细胞(DC)、髓源性抑制细胞(MDSCs)或总巨噬细胞的浸润没有观察到差异。在缺乏ADAM12+细胞的情况下,用CD8+ T细胞消耗抗体处理恢复了肿瘤生长,证实ADAM12+细胞阻断了CD8+ T细胞的抗肿瘤活性。
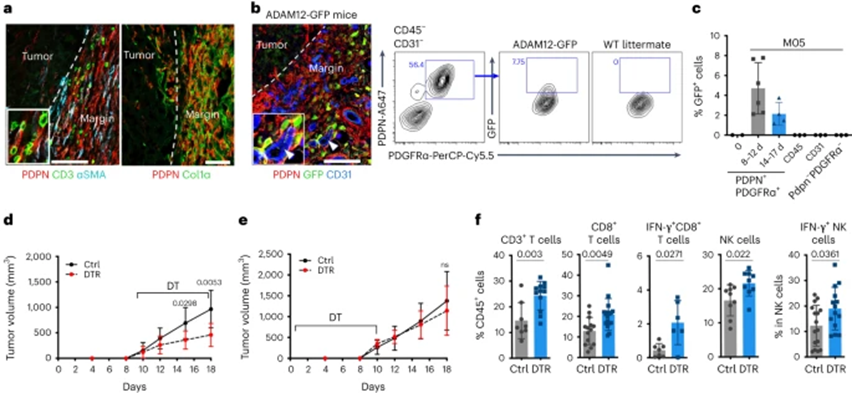
图1:ADAM12+ MSCs基因缺失恢复肿瘤免疫
2 ADAM12+ MSCs缺失使基质和血管TME正常化
与活性抗肿瘤免疫反应一致,T细胞浸润到ADAM12+细胞缺失的肿瘤中心,靠近PDPN+ CAF,PDPN+ CAFs在肿瘤内迁移并在血管附近定居(图2a、b、d)。在耗尽和对照条件下,靠近PDPN+ CAF的T细胞频率相似,与PDPN+ CAFs具有增加募集T细胞的能力相反。PDPN+ CAF的频率和绝对数量在两种条件下相似(图2c),表明CAFs对T细胞是允许的。为鉴定T细胞浸润的肿瘤中PDPN+ CAF发生的变化,对从野生型(WT)肿瘤(浸润不良)或缺乏ADAM12+细胞(浸润高度)的肿瘤中分离的PDPN+PDGFRα+细胞进行RNA-seq基因表达分析。在这两种条件下,PDPN+ PDGFRα+细胞中的差异基因表达分析确定了一些基因,包括Car9、Saa3、Lcn2、Mmp9和Mmp13,它们在缺乏ADAM12+细胞的肿瘤CAFs中显著下调(DTR与对照)(图2e)。这些基因都具有公认的抗癌作用,它们的表达通常是由缺氧诱导的。缺氧诱导的Car9(碳酸酐酶9,CAIX)通过催化二氧化碳水合为碳酸氢根离子和质子来防止胞质酸化。CAIX的表达增强了缺氧条件下的细胞存活,并增加了肿瘤微环境的酸化,这是一种主要的免疫抑制因子。与ADAM12+细胞在肿瘤缺氧和酸中毒中的作用一致,缺乏这些细胞的肿瘤显示出缺氧减少(图2f)和细胞外pH增加(图2g)。当在缺乏ADAM12+细胞的情况下恢复氧水平时,Car9在整个肿瘤中的表达显著降低(图2h)。为研究仅使肿瘤微环境的pH正常化是否足以恢复肿瘤免疫,作者用CAIX抑制剂U-104处理携带MO5黑色素瘤的WT小鼠。作者观察到,尽管CAIX抑制使细胞外肿瘤pH正常化至与ADAM12+细胞耗竭相似的水平(图2g),但抗肿瘤免疫没有恢复。此外,与缺乏ADAM12+细胞的肿瘤相比,U-104治疗的肿瘤仍然缺氧。肿瘤缺氧主要是由于功能差、肿瘤血管系统塌陷,缺乏周细胞覆盖,而周细胞覆盖是正常血管成熟和功能所必需的。与血管系统的正常化一致,缺乏ADAM12+细胞的肿瘤血管恢复了NG2+周细胞覆盖水平和宽度增加(图2i),以及ICAM1表达增加(图2j),这对白细胞粘附和跨内皮迁移至关重要,并改善了肿瘤灌注(图2k)。值得注意的是,在ADAM12+细胞缺乏的肿瘤中,PDPN+ PDGFRα+细胞中上调的基因包括了PDGF和BMP信号的调节因子(图2e),这些上调基因也参与血管成熟和正常化。
先前对小鼠黑色素瘤的单细胞RNA-seq(scRNA-seq)研究确定了三簇CAFs,称为免疫/炎症DPP4+ CD34hi基质(S1)、促结缔组织增生基质(S2)和收缩基质/周细胞(S3)。在缺乏ADAM12+细胞的肿瘤中,观察到S1细胞频率显著降低,而S2和S3的频率增加。由于S3亚群和S2亚群中CAFs表达的周细胞标记物Rgs5和Cspg4(编码NG2)水平高于S1亚群,这些数据与在ADAM12+细胞缺失的肿瘤中观察到的周细胞覆盖率增加一致(图2i)。作者进一步研究了这一机制。S1 CAFs的减少可能不是由于ADAM12+细胞的直接消融,因为大多数ADAM12+的DPP4表达较低或不表达,CD34表达中等或较低,与之前的scRNA-seq数据一致,并且占总基质细胞的一小部分(图1c)。因为ADAM12+细胞的消融减少了肿瘤缺氧(图2f),那么缺氧是否影响CAF向S1的分化。作者观察到缺氧诱导了Cd34、Dpp4、C3、Il6ra和Il6st(S1基质群体的标记基因;)在体外基质细胞中的上调,而编码广泛成纤维细胞标记物的基因(如Pdgfra和Pdpn)的表达水平不受影响。这些数据与先前的报告一致,这些报告显示炎症性CAFs在肿瘤缺氧区域富集,并进一步表明ADAM12+ MSCs的缺失通过减少肿瘤缺氧使CAFs正常化。
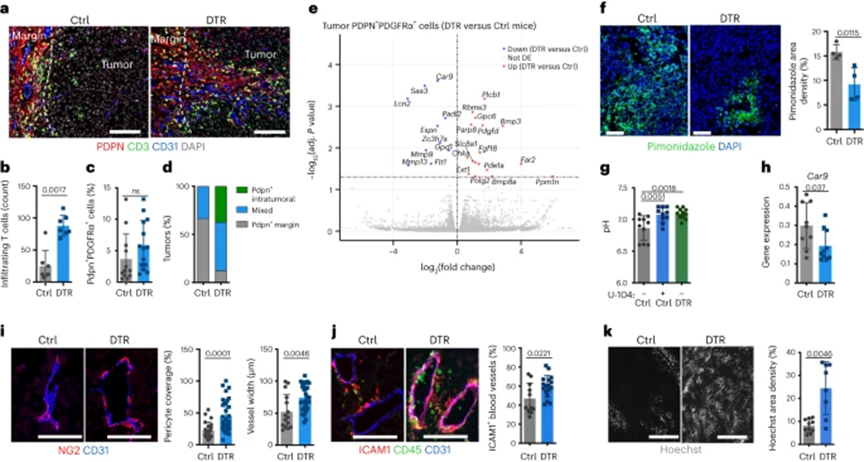
图2:ADAM12+ MSCs耗竭使基质/血管TME正常化
3慢循环ADAM12+ MSCs以TGF-β依赖的方式调节肿瘤微环境(TME)
这些数据表明,在肿瘤边缘早期发育的ADAM12+ MSCs的一小部分强烈影响血管和基质肿瘤的微环境。为研究潜在机制,在第8天对从MO5黑色素瘤中分离的ADAM12+细胞进行转录组分析。差异基因表达分析确定,与ADAM12–PDPN+ PDGFRα+细胞相比,ADAM12+ PDPN+ PDGFRα+细胞中有>700个基因显著上调(图3a)。基因集富集分析表明,ADAM12+细胞最富集通路的术语是ECM重塑、细胞增殖和分化以及炎症相关,包括生长因子受体(RTK)和细胞因子受体下游的主要信号通路,如磷酸肌醇3-激酶(PI3K)-Akt、MAPK、核因子-κB(NF-κB),肿瘤坏死因子(TNF)和JAK–STAT信号通路(图3b)。与ADAM12-细胞相比,ADAM12+细胞中与细胞代谢、蛋白质合成和DNA复制相关的几种途径下调(图3c)。在具有较低代谢的ADAM12+细胞肿瘤系中,ADAM12+细胞中参与细胞增殖、糖酵解和氧化磷酸化的基因表达下调,如Mcm2、Cdk2、Cdk15、Ldhb、Ldha和Atp5a1,而Gadd45b、Gadd45g和Gpx3表达上调,这些基因在细胞周期停滞时被诱导以响应氧化或应激损伤(图3d)。进一步表明TME内存在串扰,ADAM12+细胞表达更高水平的Pdgfra、Il1r1(编码NF-κB的激活剂)、Osmr和Il6st(gp130),这些基因编码涉及多种细胞因子受体复合物中的信号传感器,包括抑癌素(OSM)和白细胞介素-6(IL-6)(图3d)。它们还上调Il6、编码包括Cxcl1在内的趋化因子基因,以及在单核细胞募集和巨噬细胞极化中起关键作用的基因,如Ccl2、Ccl7、Csf1和Has2(图3d),以及生长停滞特异性6(Gas6)、Lgals3和Pros1,它们通过TAM(TYRO3、AXL和MERTK)酪氨酸激酶受体促进巨噬细胞胞葬。此外,ADAM12+细胞上调编码粘附分子的Icam1和ECM的促肿瘤成分,如Ctgf、Lox、Hspg2、Mmp3和Tgfbr3(图3d),表明这些细胞在TME重塑中发挥作用。与ADAM12– PDGFRα+细胞相比,ADAM12+ PDGFRα+细胞上调间充质祖细胞过表达的基因,如Ngfr、Ly6a(sca-1)、Vcam1(CD106)和PDGFRβ,但不上调Acta2(编码αSMA)或Lrrc15。这些数据表明,ADAM12+细胞是不同于αSMA+肌成纤维细胞和Lrrc15+ CAFs的血管周间充质祖细胞。
与CAF群体相比,大多数ADAM12+ MSC不表达Ki-67或不合并Edu(图3e),这与细胞处于缓慢循环状态一致。衰老标记物的缺失,如Cdkn2a(p16-INK4A)和Cdkn1a(P21),与衰老状态相反,其特征是细胞增殖和生长的不可逆停滞。因此,与对有丝分裂刺激无反应的衰老细胞不同,ADAM12+ MSC在体外给予营养时迅速恢复细胞增殖(图3f)。TGF-β在细胞周期调节中发挥重要作用。除了诱导Adam12的表达,TGF-β在从MO5肿瘤中分离的Adam12– PDGFRα+ PDPN+细胞中快速诱导编码周期阻滞蛋白Gadd45b和Gadd45g的表达,并下调参与细胞增殖的细胞周期蛋白,如Cdk6编码的细胞周期蛋白(图3g)。ADAM12+细胞过表达许多编码炎性细胞因子和生长因子受体的基因(图3d),包括Il1r1、Osmr和Il6st,表明TME内存在额外的串扰。在TME中,TAMs表达最高水平的Il1b、Osm和Pdgfc。尽管IL-1β和OSM没有诱导Adam12的表达,但IL-1β在从MO5肿瘤分离的Adam12+ MSC中诱导Cxcl1、Ccl2、Il6和Mmp3的表达,OSM快速诱导Il6、Icam1、Has2、Il1r1和Gadd45g的表达(图3h)。基质细胞中的饥饿强烈诱导了ADAM12+细胞过表达的其他因子,如Gas6,总体上表明炎症和营养缺乏的TME进一步调节了TGF-β诱导的ADAM12+细胞表型。
Tgfb1在早期肿瘤阶段被TME中的几种细胞类型上调,并且TAMs是晚期肿瘤中Tgfb1的主要产生者。由于ADAM12+细胞表达高水平的Tgfb1,和ADAM12在体外增强TGF-β信号传导,作者在体内研究了TGFBR2在MO5肿瘤中ADAM12+细胞中的作用。通过将ADAM12-tTA小鼠与tet对照的Cre(LC-1小鼠)和Tgfbr2loxP/loxP小鼠(ADAM12-tTA-CreTgfbr2小鼠)杂交,在ADAM12+细胞中产生四环素调节的Tgfbr2消融模型。由于TGF-β的表达在肿瘤早期被诱导,作者在肿瘤发生初期通过去除多西环素开始消融ADAM12+细胞中的Tgfbr2。与WT同窝出生的小鼠相比,在ADAM12-tTA-CreTgfbr2小鼠中观察到肿瘤生长的显著抑制以及T细胞浸润的增加,表明TGFBR2是ADAM12+细胞促肿瘤作用所必需的。从ADAM12-tTA-CreTgfbr2小鼠MO5肿瘤中分离的巨噬细胞表达较低水平的Vegfa,这是渗漏血管的主要诱导物,并且肿瘤血管改善了周细胞覆盖率,与血管正常化一致。因此,与从野生型肿瘤中分离的基质细胞相比,从ADAM12-tTA-CreTgfbr2小鼠MO5肿瘤中分离出的基质细胞表达更高水平的Angpt1和Pdgfrb,其通过周细胞-内皮细胞相互作用稳定血管系统,并低水平表达缺氧诱导的Car9。这些数据表明,ADAM12+细胞中的TGFBR2信号传导是其促肿瘤功能和TME改变所必需的。
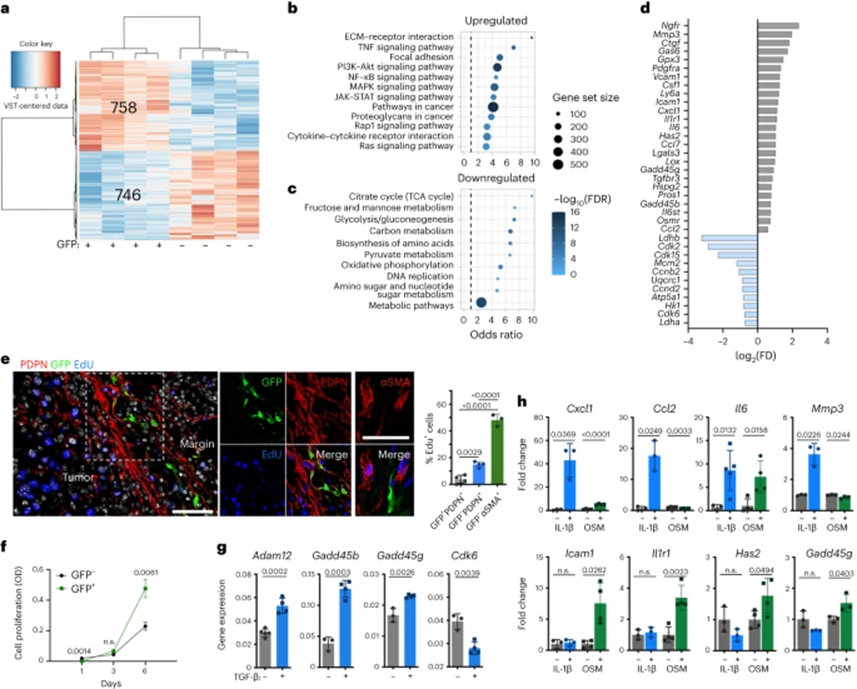
图3:慢循环ADAM12+ MSCs促进肿瘤前炎症和组织重塑
4 ADAM12+ MSCs促进巨噬细胞胞葬和极化
进一步表明ADAM12+ MSC和巨噬细胞之间存在串扰,ADAM12+细胞从早期肿瘤阶段开始在靠近CD206+ TAM的MO5肿瘤周围积累(图4a)。在较大的肿瘤中,ADAM12+细胞仍定位在肿瘤边缘,大多数(>80%)与具有磷酸化AXL的巨噬细胞非常接近(AXL-P+,图4b),这是一种对胞葬至关重要的受体,并且远离T细胞。相比之下,只有20-40%的CAFs与AXL-P+巨噬细胞相邻。巨噬细胞通过胞葬作用吞噬凋亡细胞,诱导抗炎细胞因子,如TGF-β、IL-10和前列腺素E2(PGE2),促进肿瘤发生。胞吐作用由诸如Gas6的分子增强,Gas6将凋亡细胞上的磷酸腺苷丝氨酸与TAM受体桥接。值得注意的是,与在肿瘤巨噬细胞上以较低水平表达的Mertk或Tyro3受体相比,Gas6对AXL受体具有更高的亲和力(图4c)。与作者的测序数据一致的是,PDPN+ PDGFRα+细胞在肿瘤中具有高于其他细胞类型的Gas6表达水平;ADAM12+ PDPN+ PDGFRα+细胞在RNA和蛋白水平上均具有特别高的Gas6表达水平(图4d,e)。尽管CAFs没有显示出胞葬活性,但MO5肿瘤分离的ADAM12+ MSCs(GFP+细胞)条件培养基(CM)增加了骨髓来源巨噬细胞(BMDMs)的胞葬活性,髓系细胞中的骨髓源性巨噬细胞(从LysM-CreAXLfl/fl小鼠中分离)中缺乏AXL时,这种作用被抑制(图4f)。与来自GFP-细胞CM处理的BMDM相比,GFP+细胞CM处理的渗出细胞BMDM表达更高水平的Tgfb1、Vegfa和Il10,与胞葬诱导的免疫抑制一致。体内也发生了类似的过程:ADAM12+细胞耗竭诱导AXL-P+巨噬细胞显著减少(图4g),而凋亡细胞(主要是非血管周围和非基质细胞)频率增加(图4h)。因此,与从WT肿瘤中分离的巨噬细胞相比,从缺乏ADAM12+细胞肿瘤中分离出的巨噬细胞吞噬能力降低。与WT肿瘤相比,这些数据与ADAM12+细胞缺失肿瘤中Gas6的表达降低一致,表明ADAM12+细胞是TME中Gas6的重要来源。与ADAM12+细胞在体内这一过程中的作用一致,在缺乏ADAM12+ MSC的肿瘤中,主要组织相容性复合体II(MHCII)hi CD206lo炎症巨噬细胞与MHCIIlo CD206hi TAMs的比率显著增加(图4i)。从缺乏ADAM12+细胞的肿瘤中分离的巨噬细胞表达较低水平的Tgfb1、Il10、Ptgs2(编码PGE2)和Vegfa(图4j)和上调的Light(Tnfsf14),其通过激活NK细胞、T细胞、基质细胞和恢复血管完整性来促进抗肿瘤免疫。因此,抗AXL激活抗体在缺乏ADAM12+细胞的MO5肿瘤中恢复了肿瘤进展,并将AXL-P+巨噬细胞水平增加到WT小鼠水平(图4k,l),它对WT肿瘤没有影响。总的来说,这些数据表明ADAM12+ MSC在肿瘤早期被诱导,并通过促进胞葬作用使肿瘤巨噬细胞极化为免疫抑制和促血管生成表型。
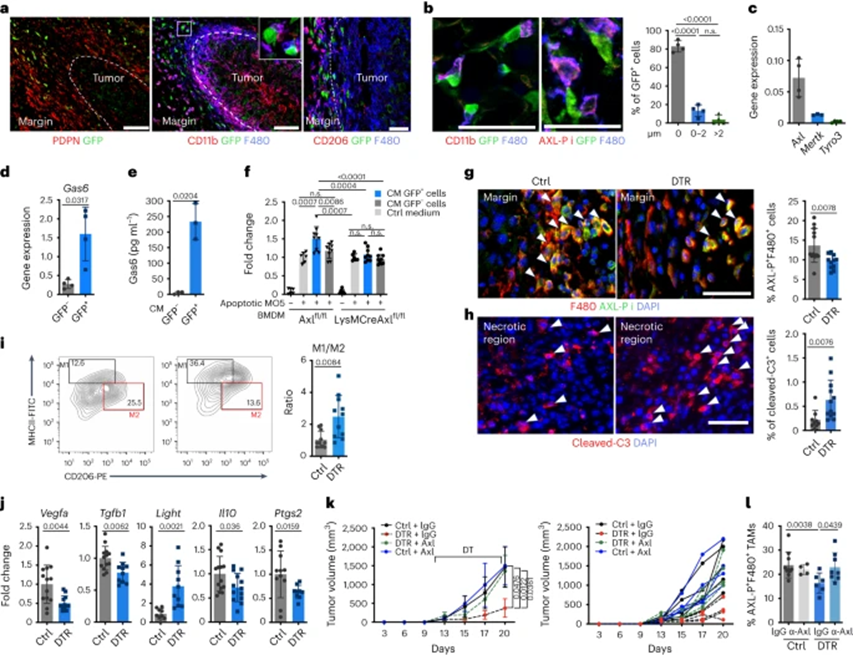
图4:ADAM12+ MSCs通过促进胞葬作用诱导免疫抑制的巨噬细胞
5肿瘤诱导的ADAM12+谱系在晚期维持
为确定ADAM12+ MSCs是否在自发发生的肿瘤中发育,将ADAM12-GFP小鼠与Rip-Tag2小鼠(一种神经内分泌胰腺肿瘤的小鼠模型)或TRAMP小鼠(前列腺腺癌的小鼠模型)。在这些模型中,SV40大T抗原表达驱动多步肿瘤进展,肿瘤分期与人类癌症相似。在这两种肿瘤中,都观察到PDPN+基质细胞和巨噬细胞在肿瘤周围区域积聚。作者观察到约1-6%的基质细胞表达ADAM12,并定位在Rip-Tag2和TRAMP肿瘤边缘的血管附近,但不在正常胰腺或前列腺中(图5a、b)。与黑色素瘤中发生的情况类似,与ADAM12-细胞相比,ADAM12+细胞上调参与细胞周期停滞、生长因子和细胞因子受体的基因,以及上调调节巨噬细胞和ECM的基因,包括Gas6、Csf1、Has2、Mmp3和间充质祖细胞标志物Ly6a和Vcam1,以及下调Mki67、Cdk1,Cdkn2a和Acta2。ADAM12+细胞表达低至中等水平的PDGFRβ,并定位于ColIV+血管BM外(图5a)。为确定肿瘤进展过程中ADAM12+ MSC在体内的命运,通过将ADAM12-tTA小鼠27与LC-1小鼠和Rosa26STOPfloxYFP报告小鼠(ADAM12tTA-CreYFP小鼠,图5c)杂交,产生ADAM12+细胞的四环素调节谱系追踪系统。在ADAM12-tTA-CreYFP小鼠中接种MO5肿瘤细胞后(多西环素维持直到肿瘤注射),作者观察到黄色荧光蛋白(YFP)+细胞(ADAM12+细胞的后代)构成了晚期黑色素瘤中约1%的基质细胞。YFP+细胞定位于肿瘤边缘、靠近血管和基质内,并表达不同水平的NG2和αSMA。与ADAM12+细胞相比,YFP+细胞下调了免疫细胞串扰所必需的几种因子,包括Ccl2、Csf1、Gas6、Lgals3、Cxcl12、Il1r1、Osmr、Il6和Angpt1,同时上调不同水平的Car9、Angpt2、Acta2和Pdgfrb。由于Car9和Angpt2编码阻断抗肿瘤免疫的蛋白质,这些数据表明ADAM12+细胞及其子代都促进免疫抑制,尽管机制不同。为确定类似的谱系是否在自发产生的肿瘤中发展,作者将ADAM12-tTA-CreYFP小鼠(图5c)与TRAMP小鼠或Rip-Tag2小鼠杂交。在前列腺和胰腺肿瘤中,观察到基质内和血管附近YFP+细胞表达中低水平的αSMA和NG2(图5e)。尽管它们靠近血管,但与周细胞相反,YFP+细胞定位在血管BM外,与分离的周细胞样细胞或血管周αSMAmid成纤维细胞一致。通过在肿瘤发生的不同阶段去除多西环素(图5d),观察到与晚期肿瘤阶段ADAM12+细胞相比,在前列腺和胰腺肿瘤的早期肿瘤阶段诱导的ADAM12+细胞具有增加基质祖细胞潜能(图5f)。多西环素处理之后,10周龄TRAMP×ADAM12-tTA-CreYFP小鼠,从第4周开始绘制命运图谱(图5g),作者分析了肿瘤早期特异性诱导的ADAM12+细胞命运。在这种情况下,在30周大的TRAMP小鼠中观察到较大前列腺腺癌肿瘤边缘中的YFP+ αSMA-或YFP+β SMAmid细胞,证明在早期肿瘤中诱导的ADAM12+细胞产生了在肿瘤进展中维持的旁癌细胞系(图5h)。
根据基因表达数据,YFP+细胞大多对增殖标记物Ki-67呈阴性(图5e,右图)。TRAMP×ADAM12-tTA-CreYFP小鼠中,在正常组织和转移到肝脏、骨骼的SV40+前列腺癌细胞的界面处也存在大量慢循环YFP+Ki-67细胞(图5i),进一步表明ADAM12+ MSC在癌转移中的作用。为研究肿瘤诱导的ADAM12+ MSC的发育起源,从发育开始对ADAM12+细胞进行谱系追踪,因为ADAM12在器官形态发生过程中表达。在ADAM12-CreYFP小鼠中,作者观察到成人健康皮肤、前列腺和胰腺中的大多数基质细胞是由胎儿ADAM12+祖细胞产生的,表明肿瘤发生重新激活了发育程序。最后,为评估ADAM12+ MSCs在自发性肿瘤模型中的作用,将ADAM12-DTR小鼠与Rip-Tag2小鼠杂交,产生Rip+DTR+和Rip+TTR-小鼠。ADAM12+细胞的耗竭诱导RIP肿瘤的显著生长抑制,显示CD3+ T细胞浸润增加。如在黑色素瘤中观察到的,PDPN+ PDGFRα+ CAF被重组,但在缺乏ADAM12+ MSC的肿瘤中仍以相似的数量存在,并且血管系统显示出周细胞覆盖率和ICAM1表达增加。这些数据表明,肿瘤发生的早期阶段诱导的慢循环ADAM12+ PDGFRα+ αSMA-血管周围MSCs产生了一个离散的间充质谱系,该谱系在晚期癌症和转移中保持并活跃。
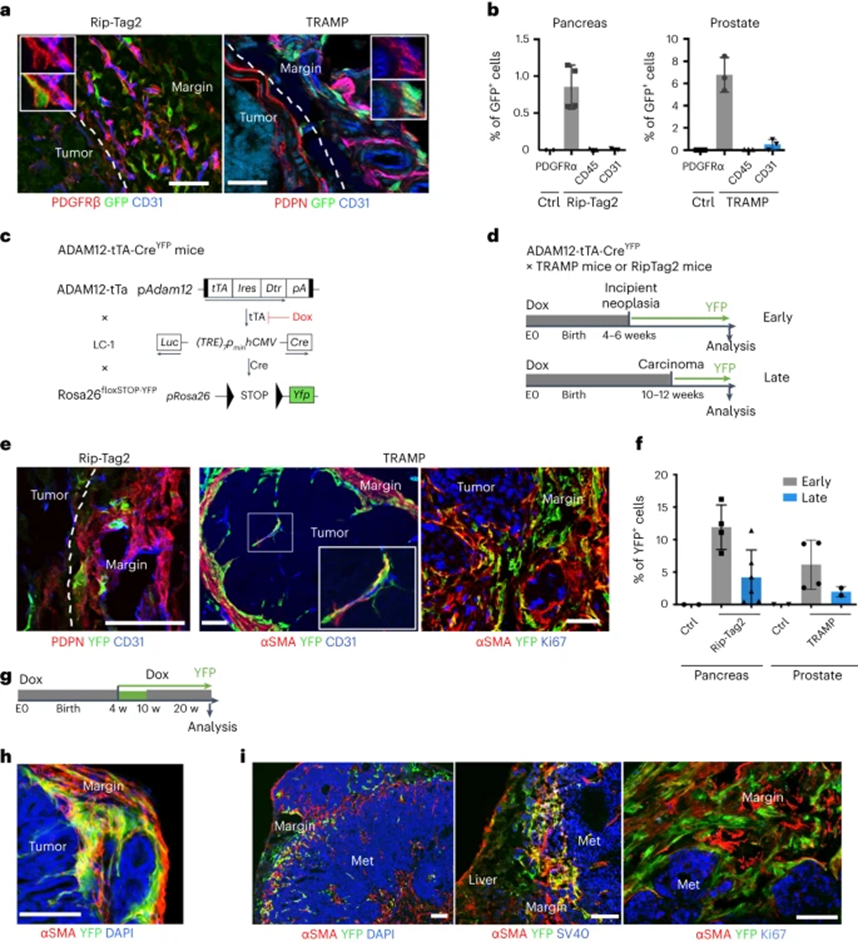
图5:肿瘤诱导的ADAM12+谱系在晚期肿瘤阶段维持
6 ADAM12能够对不同肿瘤类型的癌症患者进行分层
ADAM12在几种实体瘤中过表达,包括黑色素瘤、前列腺、乳腺、肝脏、结直肠和胰腺肿瘤,并与基质激活和不良预后有关。如在小鼠模型中观察到的,ADAM12在几种人类肿瘤中优先由基质群体表达,与之前的报道一致。在人类胰腺导管腺癌中,ADAM12的表达与“基质活化”亚型相关,后者与严重预后相关。作者进一步分析了来自皮肤黑色素瘤、胰腺导管腺癌、前列腺腺癌或结肠癌患者队列的公开数据集中ADAM12的表达。对于每个队列,通过ADAM12表达对肿瘤进行分层(ADAM12表达<中值表达与ADAM12表达>中值表达)。富集分析表明ADAM12hi肿瘤显著富集与炎症反应、缺氧和血管生成相关的基因,并且与氧化磷酸化呈负相关(图6a)。使用GO和KEGG分析进一步表明“细胞因子-细胞因子-受体相互作用”、“ECM-受体相互影响”、“组织重塑”和“伤口愈合调节”的通路富集(图6a,b)。对“细胞因子-细胞因子-受体相互作用”、“ECM-受体相互影响”、“炎症反应”和“组织重塑”通路的分析进一步确定了四个肿瘤数据集中多个共享基因(图6b-e),这表明在按照ADAM12表达分组的实体瘤中存在共享的基因表达程序。值得注意的是,与在小鼠模型中获得的数据相似,四个人类数据集中前沿常见基因包括调节或表达肿瘤巨噬细胞的基因,如AXL、CSF1、CSF1R、CD14和MSR1,以及编码OSM、IL-6和PDGF途径的细胞因子和细胞因子受体的基因,以及编码在组织重塑和血管生成中起重要作用的ECM结构和调节蛋白的基因,包括胶原蛋白、层粘连蛋白、HSPG2、HAS2、KDR和NOX4。与增加的耐药性机制一致,由ADAM12表达对人类前列腺癌的分层进一步与Gleason评分相关,该评分确定了高复发风险的高级别肿瘤。这些数据表明,在包括胰腺癌、前列腺癌和结肠癌癌症在内的几种促结缔组织增生性肿瘤中,ADAM12的表达对肿瘤患者进行了分层,这些肿瘤具有高水平的缺氧、炎症、组织重塑和先天性耐药性机制,以及与不良预后和耐药性相关的因素,如AXL。
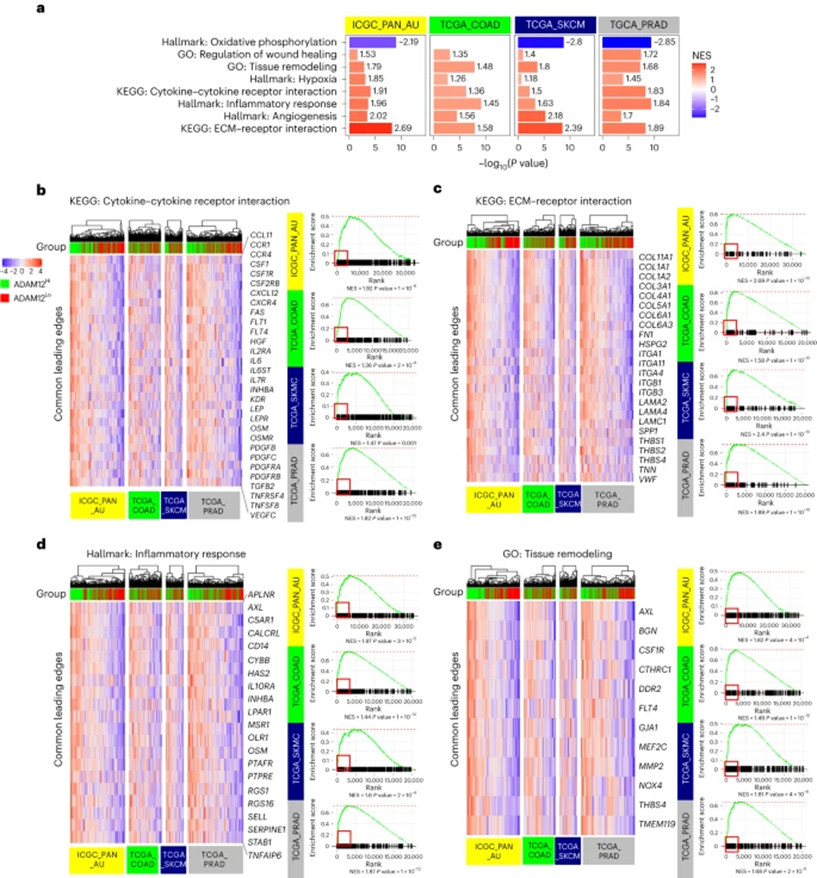
图6:ADAM12对不同肿瘤类型具有高水平缺氧、炎症和先天抵抗机制的患者进行分层
结论:
在这里,作者发现肿瘤诱导的肿瘤边缘基质细胞周期停滞协调了血管生成、组织重塑和免疫抑制,这是肿瘤进展的关键驱动因素。通过ADAM12的表达鉴定,这种代谢改变的间充质基质亚群的选择性耗竭使TME正常化,并减少肿瘤缺氧和酸中毒,诱导活化T细胞浸润和抑制肿瘤生长。作者进一步提供了直接的遗传学证据,证明ADAM12+ MSC中的TGFBR2信号传导是其促肿瘤功能所必需的。
实验方法:
小鼠肿瘤模型,免疫荧光,细胞培养,胞葬实验,qRT–PCR,RNA测序,单细胞测序
参考文献:
Di Carlo SE, Raffenne J, Varet H, Ode A, Granados DC, Stein M, Legendre R, Tuckermann J, Bousquet C, Peduto L. Depletion of slow-cycling PDGFRα+ADAM12+ mesenchymal cells promotes antitumor immunity by restricting macrophage efferocytosis. Nat Immunol. 2023 Nov;24(11):1867-1878. doi: 10.1038/s41590-023-01642-7. Epub 2023 Oct 5. PMID: 37798557; PMCID: PMC10602852.