






一种针对EAG2-Kvβ2钾通道的设计肽靶向癌细胞和神经元的相互作用来治疗胶质母细胞瘤
树突状细胞(DCs)是一种抗原呈递骨髓细胞,调节T细胞的活化、运输和功能。单核细胞来源的DCs与肿瘤抗原脉冲已广泛用于癌症治疗性疫苗接种试验,临床结果好坏参半。在这篇研究中,作者提出了一个基于小鼠或人类DC祖细胞(DCPs)的细胞治疗平台,该平台可以产生两种免疫刺激细胞因子,IL-12和FLT3L。携带细胞因子的DCPs分化为传统的i型DCs (cDC1)并抑制肿瘤生长,包括黑色素瘤和原位肝模型,而不需要抗原负载或清骨髓宿主调节。肿瘤反应涉及IL-12和FLT3L的协同作用,并与自然杀伤细胞和T细胞浸润活化、M1巨噬细胞编程和缺血性肿瘤坏死有关。抗肿瘤免疫依赖于内源性cDC1扩增和干扰素γ信号,但不需要CD8+ T细胞毒性。细胞因子武装的DCPs与抗GD2嵌合抗原受体(CAR) T细胞有效协同根除小鼠颅内胶质瘤,说明了它们在联合治疗中的潜力。本文于2023年9月发表在《Nature Cancer》,IF:22.7。
技术路线:
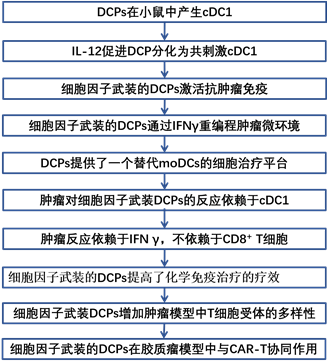
主要实验结果
1. DCPs在小鼠中产生cDC1
在小鼠和人类系统中,驻留在脑内的常见DC祖细胞(CDPs)是罕见的、有前景的cDC1前体。为了获得能够产生cDC1的细胞群,作者开发了一种从小鼠BM中体外生产cdp样细胞的方案(图1a)。这两步过程包括造血干细胞和祖细胞(HSPCs)的短期扩增,然后在促进cDC1谱系承诺的条件下进行部分分化。BM细胞在含有干细胞因子(SCF)、血小板生成素(TPO)、FMS相关酪氨酸激酶3配体(FLT3L)、IL-3、IL-6和IL-1β(扩增期)的HSPC培养基中培养2-3天。然后将漂浮细胞在含有粒细胞-巨噬细胞集落刺激因子(GM-CSF/CSF2)和FLT3L(分化期)的cDC1培养基中再培养4 - 5天。所得到的细胞培养含有类似于CDPs的细胞(CD115+、CD11b−、CD11c−、CD103−、MHCII−、CD45R/B220−、CD117/KIT−/low和CLEEC9a−),在去除谱系阳性细胞后,这些细胞的富集率约为30%至70%(图1b)。与BM驻留的CDPs不同,培养的CDPs样细胞不表达CLE9a;此外,它们缺乏CD11c和CD103,这两种蛋白在预cDC1和成熟cDC1中表达。由于它们与自然发生的CDPs相似但不相同,作者将这些细胞称为DCPs。
然后作者探索DCPs是否可以在小鼠中产生cDC1。作者使用CD45.1小鼠的BM细胞产生上述DCPs,或moDCs和成熟的cDC1样细胞,使用既定的方案。作者在没有事先骨髓消融的情况下,在基因CD45.2小鼠中静脉注射每一种DC类型(2 × 106个细胞,间隔3天),并在第二次DC剂量4天后分析受体小鼠的脾脏(图1c)。在本实验和后续实验中,流式细胞术用于识别细胞群的门控策略如图1-6所示。与接受moDCs或cDC1样细胞的小鼠相比,接受DCPs的小鼠脾细胞中供体来源的CD45.1+细胞的频率更高(图1d)。然后,作者根据早期的工作检查了脾cDC1,发现cDC1内存在大量的供体嵌合,并且在较小程度上,在接受DCPs的小鼠中,cDC2 (CD11c+CD11b+MHCII+CD8a−)(图1e)。相反,在接受moDCs或成熟cDC1样细胞的小鼠中,供体cDC1嵌合现象可以忽略不计(<0.2%)。大多数DCPs来源的细胞是cDC1, cDC2和双阴性(CD11b - CD8a -) cDCs(图1f)。作者还研究了BATF3(一种对cDC1发育至关重要的转录因子)是否在体外产生DCPs,以及它们在转移到受体小鼠后的植入和分化所必需的。从CD45.2 Batf3−/−小鼠的骨髓中可以成功建立DCPs,但在转移到CD45.1小鼠时不能移。
接下来,作者研究了皮下MC38结直肠肿瘤小鼠中供体DCPs、moDCs和cDC1样细胞的命运(图1g)。CD45.1+ DCPs来源的细胞比其他DC群体更有效地植入肿瘤和其他器官(图1)。在肿瘤中,作者在Ly6C- F4/80- CD11c+MHCII+群体中鉴定出cDC1为CD103+CD11b-细胞,cDC2为CD103- CD11b+细胞。在独立实验中,DCPs衍生的细胞分别约占肿瘤相关cDC1和cDC2的35-45%和10%(图1i)。DCPs对巨噬细胞等非cDC群体的贡献很小(<1%),并且在脾脏、肺和肝脏中也能向cDC分化,而moDCs的cDC分化能力较低。综上所述,在不需要事先的宿主调节的情况下,过继性转移的DCPs在肿瘤小鼠中有效地重建了cDC1,并在较小程度上重建了cDC2。
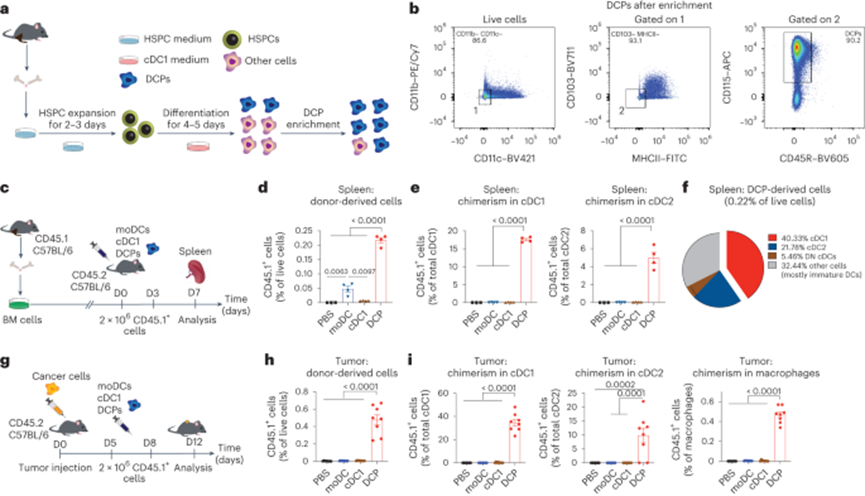
图1 DCPs能在小鼠体内高效生成cDCs
2. IL-12促进DCPs分化为共刺激cDC1
FLT3L是cDC1诱导和扩增的关键细胞因子。作者推断,在DCPs来源的细胞中强制表达FLT3L会增加肿瘤中的内源性cDC1。为此,作者构建了一个慢病毒载体(LV),表达小鼠FLT3L和绿色荧光蛋白(GFP);作为对照,作者使用仅表达GFP的LV。作者在第2天转导BM来源的HSPCs,然后测量GFP表达和FLT3L分泌。转导后的细胞在转导后第6天有效表达GF,并强烈分泌FLT3L,这是在没有外源FLT3L的情况下再培养7天的细胞条件下的酶联免疫吸附试验(ELISA)所显示。
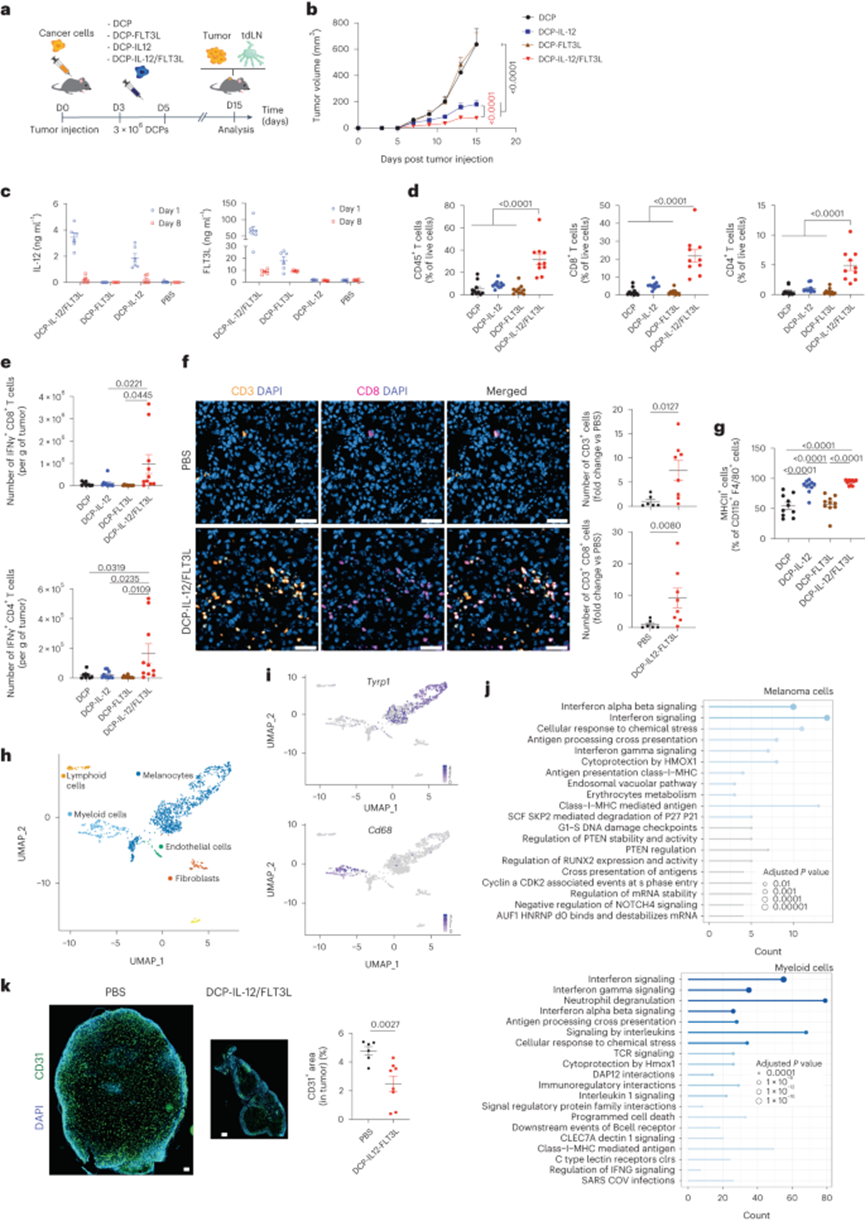
图2 携带细胞因子的DCPs激活免疫并抑制黑色素瘤生长
然后,作者使用图1a所示的方案,从CD45.1小鼠的BM中制备了对照DCPs (表达GFP的DCPs)和表达FLT3L和GFP的DCPs (以下简称DCP-FLT3L)。在本实验和随后的DCPS转移实验中,DCPS富集2小时后进行LV转导。作者在携带皮下B16F10黑色素瘤的CD45.2小鼠中接种富集的DCPs或DCP-FLT3L。与对照DCPs相比,DCP-FLT3L有效地扩大了肿瘤和脾脏中的内源性cDC。此外,DCP-FLT3L增加了CD8+和CD4+ T效应细胞(CD44+CD62L−)。因此,FLT3l武装的DCPs可能通过扩增内源性cDCs和T效应细胞启动小鼠抗肿瘤免疫。
IL-12是T细胞活化的关键细胞因子。鉴于IL-12提高了DCPs衍生的cDC1的T细胞共刺激能力,作者推断DCPs转基因表达IL-12将增强DPC-FLT3L引发的抗肿瘤免疫。作者在第2天转导脑源性HSPCs生成DCP-IL-12(表达IL-12和GFP的DCPs)和对照DCPs(仅表达GFP的DCPs)。转导后的细胞在转导后第6天稳定表达GFP, ELISA检测显示分泌IL-12。为了研究移植,作者转导了富集的CD45.1 DCPs,并在无肿瘤的CD45.2小鼠中接种了2×106个细胞。DCP-IL-12没有被反选择,保留了转基因表达,并在受体小鼠的脾脏中以预期的频率产生成熟的cDCs。
接下来,作者通过将DCPs与IL-12或FLT3L转导结合,在荷瘤小鼠中进行了DCPs转移研究。在这些实验中,IL-12与中性标记物dLNGFR(一种截断的低亲和力人神经生长因子受体)21偶联,而FLT3L与GFP偶联。作者在肿瘤攻击后的第3天和第5天给B16F10肿瘤小鼠注射了1×106 DCP-IL-12和2×106 DCP-FLT3L的混合物(图2a)。为了检验表达IL-12或FLT3L的DCPs的效果,作者将它们分别与表达GFP或dLNGFR的DCPs结合。对照小鼠接受磷酸缓冲盐水(PBS)或表达GFP或dLNGFR的DCPs。在独立实验中,与单独表达任一细胞因子的DCPs相比,DCP-IL-12和DCP-FLT3L(以下简称DCP-IL-12/FLT3L)的组合取得了更好的肿瘤控制效果(图2b),包括随访时间较长的研。在最后一次DCPS输注后的第1天至第8天,血清IL-12和FLT3L水平均急剧下降(图2c),这表明受体小鼠体内无法稳定植入产生细胞因子的DCPS。
3. 细胞因子武装的DCPs激活抗肿瘤免疫
B16F10肿瘤(包括作者研究中使用的表达OVA的变体)含有很少的T细胞浸润,并且对免疫检查点阻断反应较差。肿瘤内免疫细胞的流式细胞术分析揭示了DCPs衍生的IL-12和FLT3L之间的协同作用。与单独表达任一细胞因子的DCPs相比,DCP-IL-12/FLT3L显著增加了造血细胞、CD8+和CD4+ T细胞的肿瘤浸润(图2d)。此外,体外再刺激实验显示,DCP-IL-12/ FLT3L处理小鼠的一些肿瘤中活化的IFNγ+ T细胞比例更高(图2e)。肿瘤切片免疫荧光染色和定量分析证实了这些结果(图2f)。DCP-IL-12/FLT3L和DCP-IL-12均能增强肿瘤相关巨噬细胞(TAM) MHCII的表达(>90%;图2g和扩展数据图5d),表明获得免疫刺激表型。最后,在DCP-IL-12/ FLT3L处理的小鼠中,肿瘤引流淋巴结(tdLNs)中CD44+CD62L−T效应细胞的相对丰度更高,表明激活了全身T细胞反应。这些结果表明,IL-12/ FLT3L武装DCPs在T细胞贫乏的黑色素瘤模型中促进广泛的免疫反应。
4. 细胞因子武装的DCPs通过IFNγ重编程肿瘤微环境
然后,作者对整个B16F10肿瘤进行了单细胞RNA测序(scRNA-seq)分析,作者在第二次DCPs给药后6天进行了分析。由于黑素小体在黑色素瘤制备中大量存,作者只能准确地识别主要的细胞簇(图2h)。尽管如此,作者观察到与载体治疗的肿瘤相比,DCP-IL-12/ FLT3L治疗的肿瘤的癌症和髓细胞中IFNγ和i型IFN信号通路以及其他免疫反应途径(例如抗原递呈)的激活,如根据Reactome和Hallmark对最不受管制的生物过程的无监督排名所示(图2i,j)。正如预期的那样,IFNγ仅在淋巴细胞簇中可检测到表达。结合上述基于流量的数据,scRNA-seq分析强烈表明,DCP-IL-12/FLT3L激发了IFNγ反应,至少部分地通过对黑色素瘤和骨髓细胞的影响,有助于限制肿瘤生长。此外,在DCP-IL-12/FLT3L治疗的黑色素瘤中存在抗血管生成反应(图2k),这可能涉及IFN-γ27的直接血管修剪作用和间接机制;例如,通过m1编程(血管静压)TAMs28。
5. DCPs提供了一个替代moDCs的细胞治疗平台
大多数载抗原DC的临床前和临床实验使用的是moDCs。然后,作者研究了IL-12和FLT3L的转基因表达是否会赋予moDCs在缺乏体外抗原负载的情况下扩增T细胞和控制肿瘤生长的能力。作者将1 × 106 DCP-IL-12或moDC-IL-12和2×106 DCP-FLT3L或moDC-FLT3L的混合物给予B16F10肿瘤小鼠,在肿瘤攻击后的第3天和第5天(图3a)。DCP-IL-12/FLT3L实现肿瘤稳定,而moDC-IL-12/FLT3L仅延缓肿瘤生长(图3b)。在接受DCP-IL-12/FLT3L的小鼠中,这一结果与肿瘤中CD8+ T细胞的浸润和活化显著增加(图3c)以及TDLN中cDCs和T效应细胞(CD44+CD62L−)的扩增相关。此外,经体外再刺激,DCP-IL-12/ FLT3L处理的小鼠肿瘤中表达IFNγ、颗粒酶B (GZMB)和肿瘤坏死因子(TNF)的CD8+ T细胞比例更高(图3d)。虽然DCP-IL-12/FLT3L和moDC-IL-12/FLT3L均可适度增加肿瘤中CD4+ T细胞的浸润和活化,但只有DCP-IL-12/FLT3L可增强tam中MHCII的表达。总体而言,DCP-IL-12/ flt3l处理小鼠的肿瘤细胞组成以T细胞为主,几乎占活细胞的三分之二(图3e)。这一结果可以解释DCP-IL-12/FLT3L治疗后观察到的普遍IFNγ特征和标记的M1编程。
上述研究使用了以1:2比例表达IL-12或FLT3L的混合dc。为了探索作者平台的多功能性,作者从一个双胞LV中共表达了这两种细胞因子,即在同一个细胞中,这代表了一种更适合临床翻译的策略。DCP-IL-12/FLT3L和moDC-IL-12/FLT3L在体外共表达IL-12和FLT3L,并在B16F10模型中表现出抗肿瘤活性(图3f、g)。在涉及LV分裂转导的研究中观察到,尽管moDC-IL-12/FLT3L剂量加倍可改善肿瘤控制,但DCPs比moDCs更有效。总的来说,这些临床前结果表明,细胞因子武装的DCPs为抗原不可知的DC治疗应用提供了一种替代moDCs的策略。
然后,作者在MC38模型中检测DCP-IL-12/FLT3L,其特征是免疫抑制TAMS大量浸润。MC38荷瘤小鼠的处理方法与上面图3a所示的黑色素瘤研究相同。DCP-IL-12/FLT3L实现了实质性的MC38肿瘤控制(图3h),促进免疫细胞浸润(图3i),诱导TAM获得M1样表型(图3j,k)。此外,DCP-IL-12/FLT3L强烈扩增tdLNs中的cDCs和CD44+CD62L−T效应细胞(图31),这与B16F10黑色素瘤模型中的发现一致。
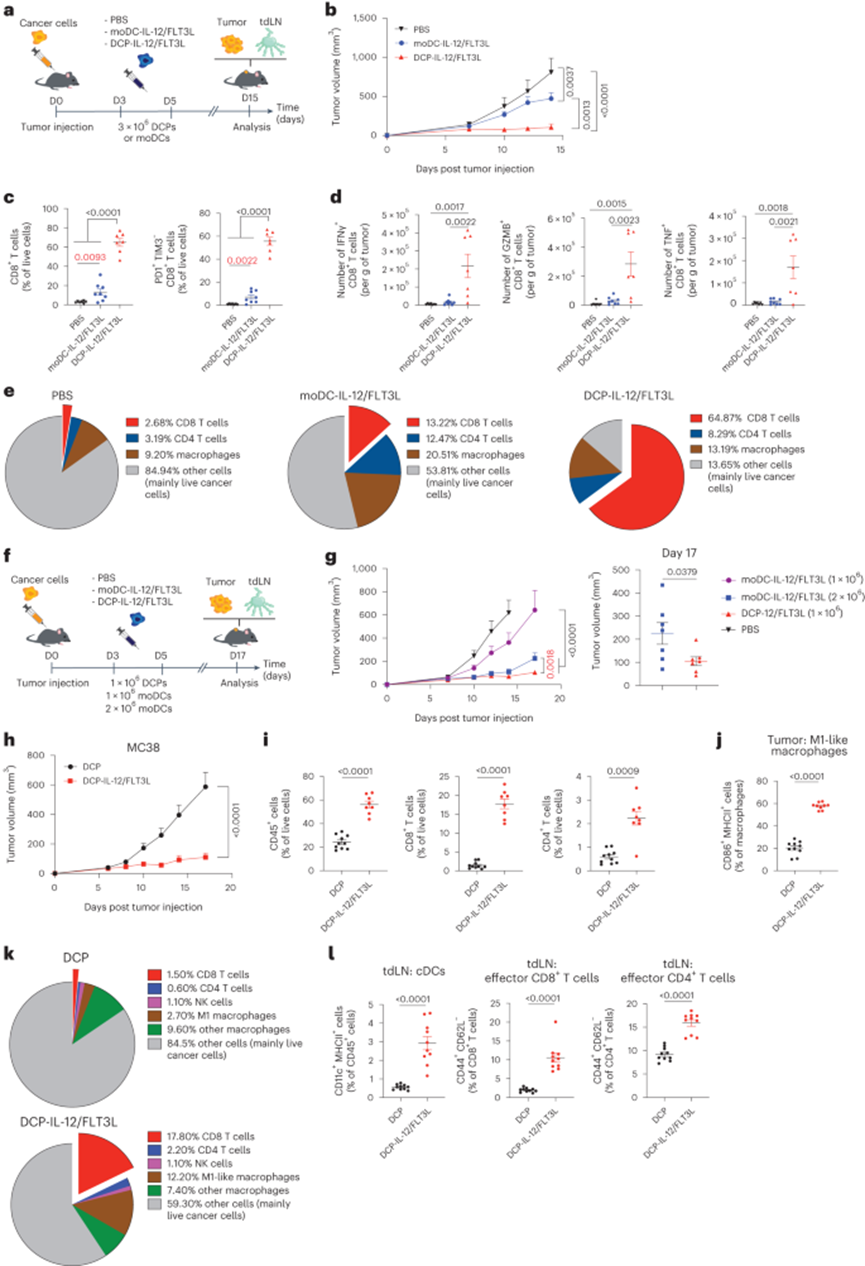
图3 DCPs为moDCs提供了一种有效的细胞因子递送平台
6. 肿瘤对细胞因子武装DCPs的反应依赖于cDC1
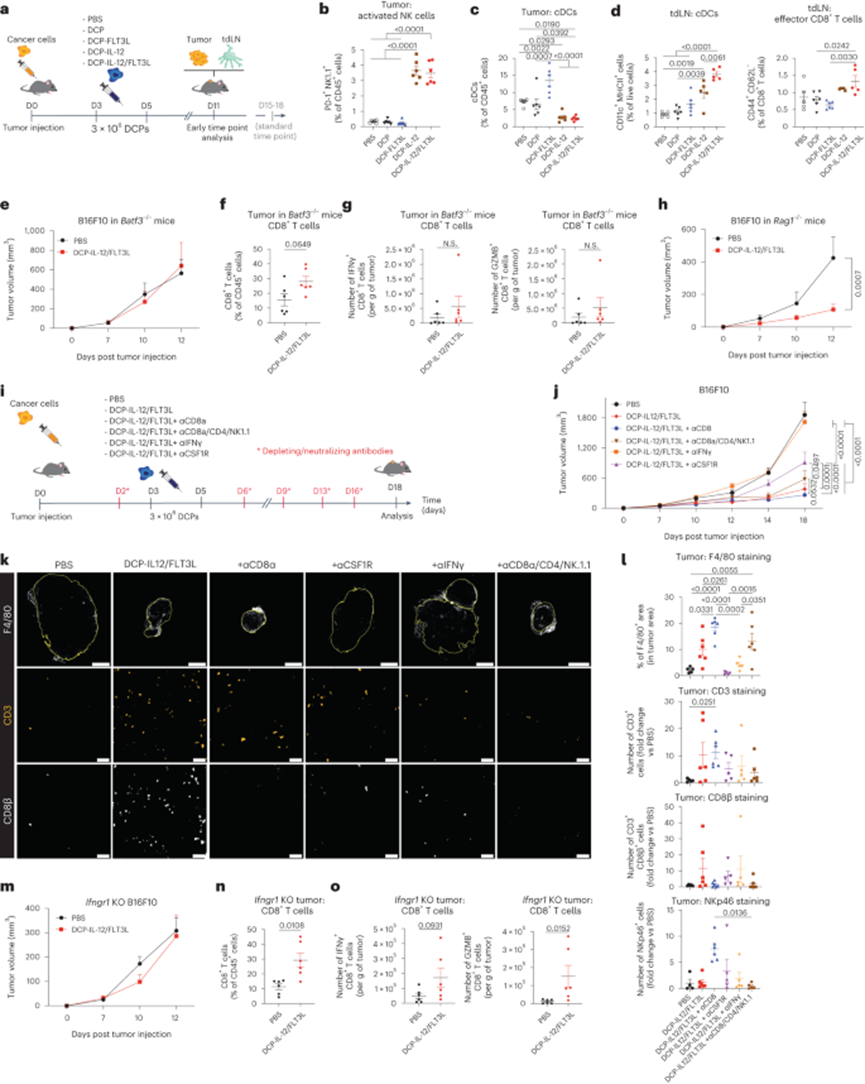
图4 肿瘤对细胞因子DCPs的反应是 cDC1 和 IFNγ 依赖性的,但不需要 CD8+ T 细胞
为了探索肿瘤对DCP-IL-12/FLT3L反应的潜在机制,作者研究了DCPs转移后早期时间点的B16F10肿瘤(第二次DCPs剂量后6天;图4)。在肿瘤发展的早期阶段,很少有造血细胞和CD8+T细胞浸润,DCP-IL-12或DCP-IL-12/FLT3L仅适度增加了造血细胞和CD8+T细胞浸润。然而,后一种治疗显著增加了肿瘤中活化的自然杀伤(NK)细胞的丰度(图4b)。因此,DCP-IL-12/FLT3L的早期肿瘤反应可能主要涉及IL-12对NK细胞的依赖作用。有趣的是,DCP-IL-12和DCP-IL-12/FLT3L表现出双重作用,减少肿瘤中的cDCs,同时增加tdLNs中的cDCs,包括CD11clow/+MHCII+/高迁移cDCs(图4c、d)。这种反应伴随着tdLNs中CD44+CD62L−T效应细胞的适度增加。相反,与tdln相比,DCP-FLT3L诱导肿瘤内cdc的增加更为明显。这些结果支持了DCPs衍生的FLT3L直接促进内源性cDCs在肿瘤中的初始扩张的假设,而IL-12通过NK细胞衍生的IFNγ诱导它们从肿瘤微环境(TME)迁移到tdLN。这种迁移将使cDC能够启动tdLN中的T细胞启动。
然后,作者探索内源性cDC1和T细胞是否需要对DCP-IL-12/FLT3L的肿瘤抑制反应。在Batf3−/−受体小鼠中缺乏内源性cDC1完全否定了DCP-IL-12/FLT3L的治疗活性(图4e),并且无法在B16F10肿瘤中引起强大的T细胞浸润和激活(图4f,g),这表明细胞因子修饰的DCPs通过内源性cDC1激活抗肿瘤免疫。令人惊讶的是,DCP-IL-12/FLT3L在缺乏成熟T细胞和B细胞的Rag1−/−小鼠中也有效(图4h)。在Rag1−/−小鼠的B16F10肿瘤中,作者观察到活化的PD-1+ NK细胞的比例大大增加,这可能至少部分解释了抗肿瘤作用的持久性。B16F10肿瘤接种于Batf3−/−、Rag1−/−和野生型小鼠的免疫细胞组成。这些结果使作者推测,DCP-IL-12/FLT3L的抗肿瘤活性依赖于内源性cDC1,而不需要T效应细胞的杀肿瘤功能。
7. 肿瘤反应依赖于IFN γ,不依赖于CD8+ T细胞
为了进一步了解T细胞参与肿瘤对DCP-IL-12/FLT3L的反应,作者在B16F10肿瘤小鼠中进行了细胞耗尽和细胞因子中和研究(图4i)。与Rag1−/−小鼠的研究结果一致,消除CD8+ T细胞并不影响肿瘤对DCP-IL-12/FLT3L的反应,值得注意的是,同时消除CD8+ T细胞、CD4+ T细胞和NK1.1+ NK细胞只能适度地挽救肿瘤生长(图4j)。相比之下,中和IFNγ完全恢复肿瘤生长,而使用集落刺激因子1受体(CSF1R)抗体消耗TAM部分恢复肿瘤生长。值得注意的是,CSF1R不影响肿瘤和tdLN中的cDC数量,这与CSF1R−/−小鼠的研究结果一致。鉴于流式细胞术分析捕获了相对细胞比例,作者使用非竞争抗体对肿瘤切片进行免疫荧光染色以获得定量数据(图4k, 1)。DCP-IL-12/FLT3L增加F4/80+ TAM,这一反应被CSF1R阻断和IFNγ中和所消除,但不被T细胞和NK细胞消除所消除。此外,DCP-IL-12/FLT3L增加了CD8+和总CD3+ T细胞,但消除CD8+ T细胞并没有减少总CD3+ T细胞数量,提示CD3+CD8−T细胞代偿性浸润肿瘤。有趣的是,CD8+T细胞消除增加了NK细胞和tam,即使在CD8+和CD4+ T细胞和NK细胞联合消除后,它们仍然升高。在一项独立研究中,单独消除CD4+ T细胞或NK1.1+ NK细胞不会损害肿瘤对DCP-IL-12/FLT3L治疗的反应,并且破坏CD4+ T细胞与总CD8+ T细胞和活化(IFNγ+或GZMB+) CD8+ T细胞的代偿性增加有关。总的来说,这些发现表明B16F10肿瘤对DCP-IL-12/FLT3L的反应严格依赖于IFN Γ,涉及多种IFN Γ产生细胞,并且可能至少部分依赖于IFN Γ刺激的M1样TAM的抗肿瘤活性。
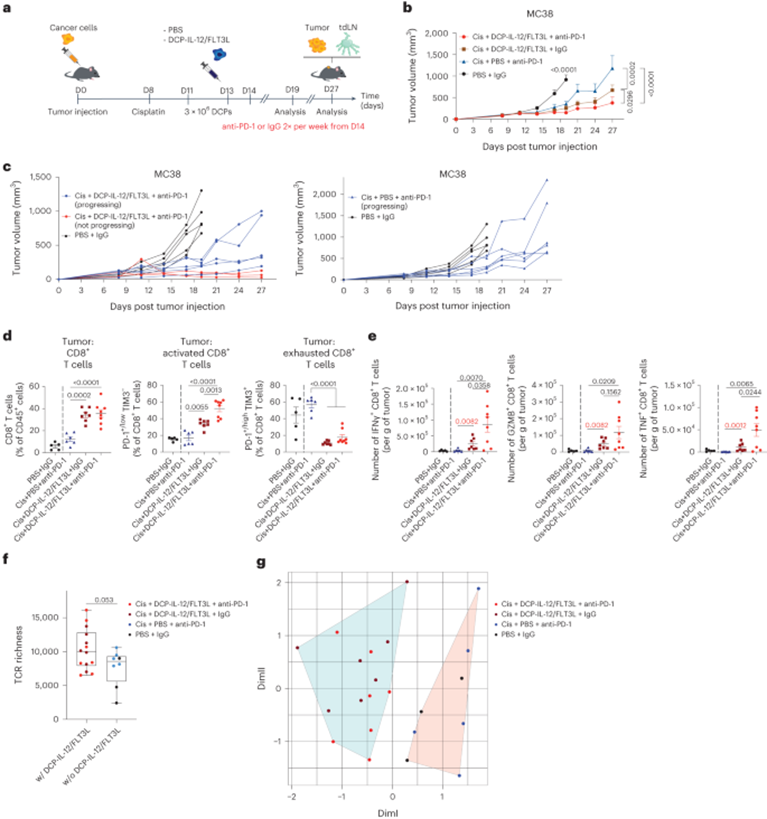
图5 细胞因子武装DCPs在结直肠癌模型中提高了顺铂和PD-1阻断的疗效
最后,为了探索IFNγ对B16F10黑色素瘤细胞的潜在直接影响,作者产生了ifngr1敲除的B16F10细胞。尽管观察到肿瘤内CD8+ T细胞浸润和活化增强(图4n,o),但消除癌细胞对IFNγ的反应性足以否定DCP-IL-12/FLT3L在小鼠中的治疗活性(图4m)。综上所述,作者的研究结果表明IFNγ是肿瘤抑制的关键介质,并强烈表明IFNγ的产生,而不是直接的CD8+ T细胞毒性,是DCP-IL-12/FLT3L治疗反应所必需的。
8. 细胞因子武装的DCPs提高了化学免疫治疗的疗效
B16F10和MC38肿瘤表现出快速的生长动力学,这使得晚期肿瘤治疗干预的评估复杂化。为了减缓MC38肿瘤的生长,作者用单剂量顺铂(一种用于结直肠癌治疗的化疗药物)对MC38荷瘤小鼠进行预处理。随后在第11天和第13天注射DCP-IL-12/FLT3L,从第14天开始注射PD-1阻断抗体,每周两次(图5a)。虽然顺铂和抗PD-1联合使用延迟了肿瘤生长,但在顺铂中加入DCP-IL-12/FLT3L可改善抗肿瘤反应,PD-1阻断可进一步改善这种反应(图5b)。联合治疗(顺铂,DCPs,抗PD-1)导致8只小鼠中的3只肿瘤消退或稳定(第27天和第11天),而其他组中所有肿瘤进展(图5c)。DCP-IL-12/FLT3L联合顺铂增加了造血细胞、CD8+和CD4+ T细胞在肿瘤内的浸润,不依赖于PD-1阻断(图5d和扩展数据图8b)。值得注意的是,大多数T细胞表现出非耗尽的激活表型。此外,体外再刺激实验显示,在接受完整治疗方案的小鼠肿瘤中,CD8+ T细胞中IFNγ、GZMB和TNF的表达增强,CD4+ T细胞中IFNγ的表达升高(图5e),这可能解释了PD-1阻断的附加益处。相反,用顺铂和抗PD-1治疗的小鼠肿瘤内CD8+或CD4+ T细胞未升高,主要表现为衰竭表型。这些数据表明,细胞因子武装的DCPs提高了结直肠癌模型的化学免疫治疗效果。
9. 细胞因子武装DCPs增加肿瘤模型中T细胞受体的多样性
作者通过T细胞受体β (TCRβ)的批量测序来检测MC38肿瘤中的T细胞多样性。作者观察到接受DCP-IL-12/FLT3L的肿瘤的T细胞库具有更高多样性的趋势,如更多的独特克隆型所示(图5f)。有趣的是,如无监督k均值聚类所示,接受DCP-IL-12/FLT3L治疗的小鼠肿瘤中的T细胞在其TCR中的V基因使用方面具有显著的相似性(图5g)。这些发现表明,DCP-IL-12/FLT3L促进T细胞的扩增,并对MC38肿瘤相关抗原具有共同的特异性。
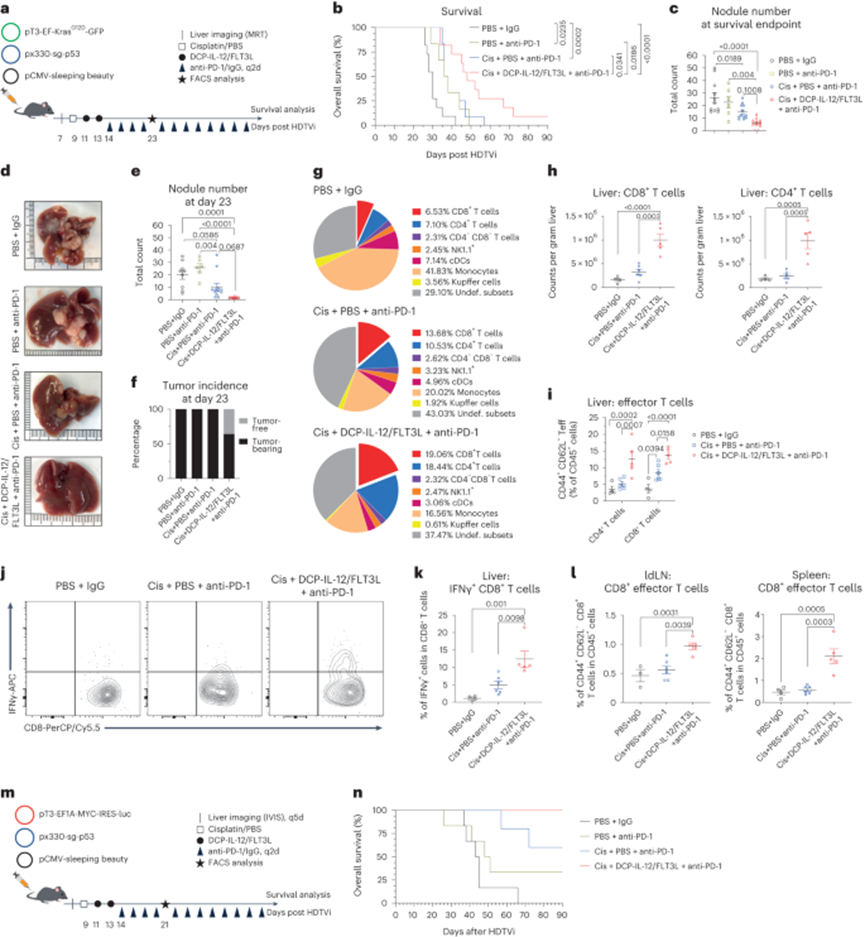
图6 细胞因子武装的DCP在两种基因工程肝癌模型中均有效
作者探索DCPs是否在两种通过流体动力尾静脉注射(HDTVi)致癌质粒获得的基因工程肝癌模型中也有效。在第一个模型中,在肝细胞中激活KrasG12D癌基因并缺失Trp53可诱导具有肝细胞癌和胆管癌特征的多灶性肝脏肿瘤。KrasG12D; Trp53−/−肿瘤建立了免疫抑制和几乎免疫荒漠的TME,并表现出侵袭性生长模式,小鼠中位生存期约为30天。单用抗PD-1、顺铂联合抗PD-1或DCP-IL-12/FLT3L联合顺铂和抗PD-1治疗肿瘤启动小鼠(图6a)。与其他治疗相比,DCP-IL-12/FLT3L显著延长了生存期(图6b)。值得注意的是,尽管从肿瘤诱导到终止的平均时间较长,但这些小鼠的大肿瘤较少(图6c)。
在两项独立研究中,作者分析了固定时间点(肿瘤发生后第23天)的肝脏。此时,三联组小鼠的宏观肝脏肿瘤数量大幅减少,11只小鼠中有4只无肿瘤(图6d-f),与生存研究结果一致。肝实质免疫荧光染色显示,与其他治疗相比,大多数接受DCP-IL-12/FLT3L治疗的小鼠的CD8+和CD4+ T细胞密度增加。作者还在分析的同一时间点(第23天)用流式细胞术检测免疫细胞参数。DCP-IL-12/FLT3L增加了肝实质中的CD8+和CD4+ T细胞,无论是CD45+造血细胞的比例(图6g)还是绝对细胞计数(图6h)。此外,DCP-IL-12/FLT3L增加了CD44+CD62L−CD8+和CD4+ T效应细胞(图6i)和IFNγ+ CD8+ T细胞(图6j,k)。在肝引流淋巴结和脾脏中也观察到更高比例的CD44+CD62L−CD8+ T效应细胞(图6l)。
然后,作者采用了基于HDTV的Myc驱动和Trp53缺失肝癌模型,该模型比KrasG12D; Trp53−/−模型发生的肿瘤更少。Myc; Trp53−/−肿瘤具有肝细胞癌的特征,具有功能失调的DC,并且对免疫检查点阻断具有抗性。单用抗PD-1、顺铂联合抗PD-1或DCP-IL-12/FLT3L联合顺铂和抗PD-1治疗肿瘤启动小鼠(图6)。在该模型中,DCP-IL-12/FLT3L达到了100%的生存率,优于其他治疗组的生存率(图6n)。此外,在固定时间点(肿瘤发生后第21天)分析的独立小鼠队列显示,DCP-IL-12/FLT3L组的6只小鼠中有5只没有宏观肿瘤的证据,而其他组中至少有50%的小鼠有肿瘤。流式细胞术分析显示,在接受DCP-IL-12/FLT3L治疗的小鼠中,CD4+(而非CD8+) T细胞比例增加;这种反应与肝引流淋巴结和脾脏中CD4+和CD8+ T效应细胞比例升高有。总之,在两种侵袭性肝癌模型中,细胞因子武装DCPs通过诱导产生IFN γ的T细胞、降低肿瘤多样性和延长生存期来改善肿瘤对顺铂和抗PD-1的反应。
10. 细胞因子武装的DCPs在胶质瘤模型中与CAR-T协同作用
CAR-T是具有工程肿瘤特异性的T细胞。虽然CAR-T可以识别并杀死表达靶向抗原的癌细胞,但其在实体肿瘤中的治疗效果受到抗原异质性和免疫抑制TME的限制。作者使用了侵袭性SB28小鼠胶质瘤模型,它概括了人类胶质母细胞瘤的关键特征,免疫沉默,对免疫检查点阻断无反应。作者使用了针对GD2的CAR-T, GD2是一种在人类胶质瘤亚群中表达的双胞脂苷,也是一种经临床验证的CAR-T靶标。GD2+ SB28胶质瘤细胞是通过用编码GD2合成酶、GD2S和GD3S的lv转导亲本细胞系产生的。抗GD2 CAR-T在体外有效杀伤GD2+细胞,但不杀伤GD2 - SB28细胞。
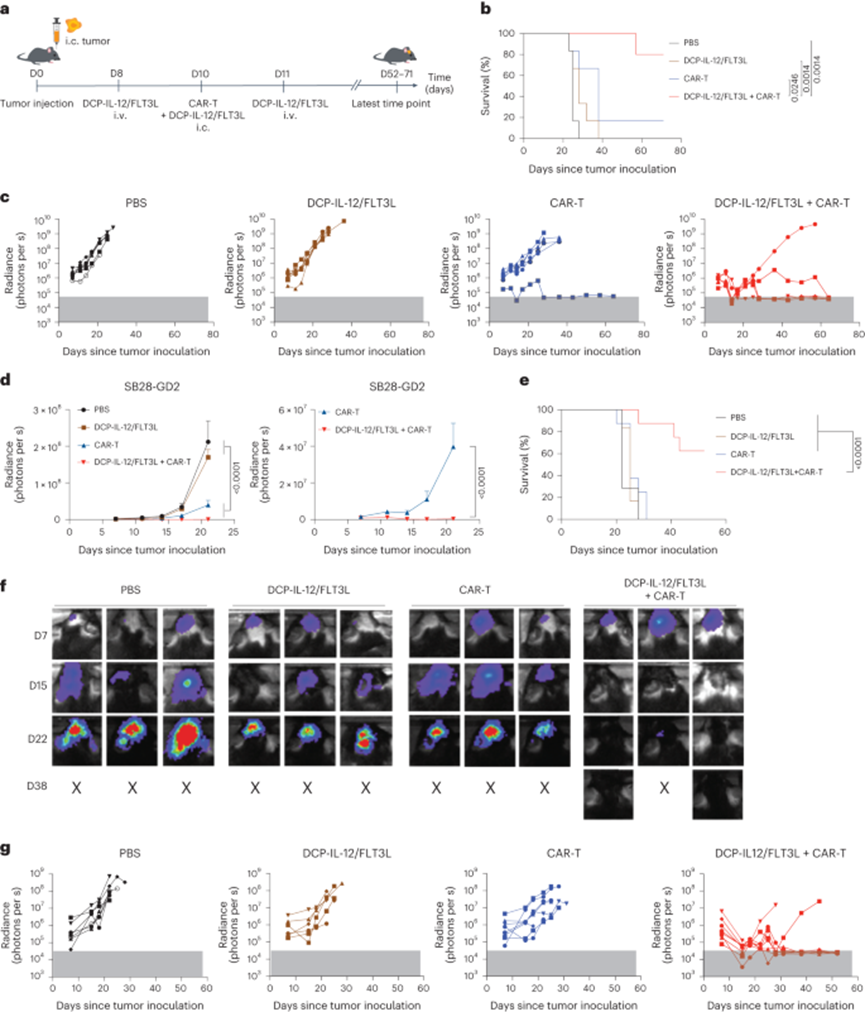
图7 细胞因子武装的DCP在两种基因工程肝癌模型中均有效
作者在小鼠脑内接种SB28-GD2细胞,并用DCP-IL-12/FLT3L、抗GD2 CAR-T或两者的组合处理它们(图7a)。通过纵向实时成像分析监测肿瘤进展。接受DCP-IL-12/FLT3L和CAR-T联合治疗的小鼠存活时间明显长于单独接受任何一种细胞治疗的小鼠(图7b)。虽然DCP-IL-12/FLT3L或CAR-T单药治疗中度延迟肿瘤进展,但CAR-T队列中除一只小鼠外,所有小鼠均出现进展性疾病(图7c,d)。相反,联合治疗在5只小鼠中有4只小鼠肿瘤消退,直到研究结束(肿瘤接种后第71天)仍无肿瘤。
上述研究使用了表达IL-12或FLT3L的DCPs混合物。然后,作者使用双频IL-12/FLT3L LV单导的DCPs重复了胶质瘤研究。与上述结果一致,DCP-IL-12/FLT3L + CAR-T在8只小鼠中根除了6只小鼠的肿瘤(1只小鼠在无肿瘤时被杀死),而其他组的所有小鼠都出现了进行性疾病(图7e-g)。死后大脑免疫荧光染色显示,通过活体成像评估为无肿瘤的小鼠中未检测到胶质瘤细胞;有趣的是,大量的CD3+ T细胞浸润持续存在于肿瘤完全消退的部位。因此,DCP-IL-12/FLT3L与GD2特异性CAR-T协同作用可根除小鼠大部分颅内胶质瘤。
小鼠DCPs的临床前疗效促使作者测试人类脐带血CD34+ HSPCs是否支持DCPS分化。在1周内,在FLT3L、IL-3、IL-6、TPO和小分子UM729存在下培养的CD34+细胞显著扩增(图8 e),并获得更分化的祖细胞状态,包括常见的髓系祖细胞、粒细胞-单核细胞和DC祖细胞、单核细胞-DC祖细胞(MDPs)、CDPs和前DCs。虽然表达CDP标记物的细胞频率可以忽略不计(<0.1%),但培养的细胞含有相当大比例(约7%)的MDPs。鉴于MDPs是单核抗原呈递细胞(APCs)的前体,包括CDPs、cDC1和cDC2,作者研究了含有MDPs的细胞群(鉴定为Lin-CD34+CD115+,暂时称为DCPs)产生cDCs的能力。在第7天,Lin-CD34 +CD115+ DCPs表达了CDPs和MDPs之间共享的标记(例如,CD117/KIT, CD135和CD45RA;图8 b)。这些细胞可以从脐带血和动员外周血(MPB)中获得,MPB是CD34+ HSPCs41的临床来源。然而,脐带血比MPB产生更多的DCPs(图8c,d)。脐带血和MPB衍生的DCPs都可以被表达dLNGFR的LV有效转导(图8e),这表明LV转导在该细胞群中是可行的。
为了研究Lin-CD34+CD115+ DCPs的APC分化能力,作者从7天脐带血或MPB培养中分离出Lin-CD34+CD115+ DCPs,并在cDC培养基(含FLT3L、GM-CSF、SCF和IFNα)中培养。作为对照,作者模拟了第7天的细胞,其中大多数是CD115−。1周后(第14天),Lin-CD34+CD115+细胞有效地分化为APC,包括单核细胞、cDC1、cDC2和未成熟的DC,其他细胞类型的贡献较小,而是在模拟分类细胞培养中扩增(图8f)。这些结果表明Lin-CD34+CD115+细胞可以作为人DCPs发挥作用。
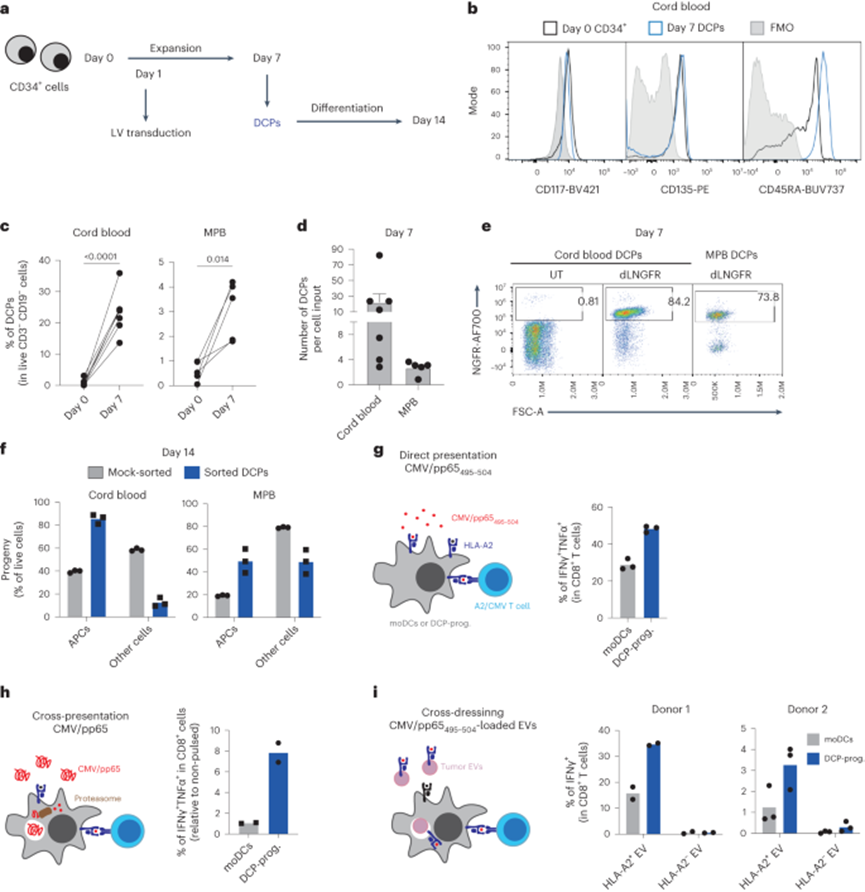
图8 人HSPC是具有抗原呈递能力的DCP的来源
接下来,作者评估了人类DCPS衍生细胞(称为DCPs后代)和moDC的抗原呈递能力。作者使用巨细胞病毒(CMV)蛋白PP65和HLA - A2限制性CMV特异性T细胞。作者研究了三种抗原呈递途径:(1)呈递PP65495-504肽负载的HLA-A2,模拟直接呈递;(2)交叉呈递由天然PP65蛋白内源性加工而成的PP65495-504肽;(3)细胞外囊泡携带PP65495-504 HLA-A2对DC进行抗原交叉修饰。T细胞与先前暴露于PP65495-504肽、PP65蛋白或装载PP65495-504的细胞外囊泡的DCPS子代或moDCs共培养,分别测定直接呈递、交叉呈递和交叉呈递。对于变装,作者使用从HLA-A2+或阴性的人类黑色素瘤细胞中分离的细胞外囊泡和从HLA-A2阴性供者获得的DC,前提是HLA-A2阴性的细胞外囊泡不会通过变装激活HLA-A2限制性T细胞。在每种情况下,DCPS后代比moDCs更有效地激活cmv特异性T细胞,尽管T细胞的激活程度因供体而异(图8g – 1)这些结果表明,人类DCPs,鉴定为Lin-CD34 +CD115+细胞,可以分化为具有抗原呈递能力的细胞后代。
结论
这一发现表明,DCPs部署IL-12可能直接增强CAR-T细胞。或者,DCPs可能协调IFN γ依赖的内源性免疫反应,消除逃避CAR-T细胞识别和杀死的癌细胞。这些结果,再加上开发人类DCPs样细胞的可行性,激发了基于抗原不可知的DCPs治疗的进一步临床前研究。
实验方法:
逆转录病毒载体的设计和生产,细胞培养,小鼠骨髓细胞的分离,小鼠DCPs的生成, DCPs的分化,小鼠moDCs和CDC1样细胞的生成,OT-I和OT-II T细胞与载抗原cDC1样细胞共培养,小鼠DCP和moDCs与LVs的转导,CAR-T细胞产生,CAR-T细胞杀伤试验,流式细胞术分析,免疫荧光染色,人T细胞刺激试验,scRNA-seq,整体TCR测序。
参考文献:
Dong, W., Fekete, A., Chen, X., Liu, H., Beilhartz, G. L., Chen, X., Bahrampour, S., Xiong, Y., Yang, Q., Zhao, H., Kong, T., Morioka, M. S., Jung, G., Kim, J. E., Schramek, D., Dirks, P. B., Song, Y., Kim, T. H., He, Y., Wanggou, S., … Huang, X. (2023). A designer peptide against the EAG2-Kvβ2 potassium channel targets the interaction of cancer cells and neurons to treat glioblastoma. Nature cancer, 4(10), 1418–1436. https://doi.org/10.1038/s43018-023-00626-8
