






hsa_circ_0007919增强胰腺导管腺癌DNA损伤应答,促进吉西他滨耐药
circRNAs在癌症和化疗耐药的发生和发展中发挥着重要作用。DNA损伤修复有助于癌细胞的增殖和对化疗诱导的细胞凋亡的抵抗。然而,circRNA在DNA损伤修复调控中的作用需要澄清。我们在耐GEM的PDAC组织和细胞中发现了高表达的circRNA hsa_circ_0007919。hsa_circ_0007919的高表达与PDAC患者较差的总生存期(OS)和无病生存期(DFS)相关。Hsa_circ_0007919以依赖LIG1的方式抑制GEM诱导的DNA损伤、DNA断裂积累和细胞凋亡,维持细胞存活。机制上,hsa_circ_0007919招募FOXA1和TET1降低LIG1启动子的甲基化并增加其转录,进一步促进碱基切除修复、错配修复和核苷酸切除修复。最后,我们发现GEM增强了QKI与hsa_circ_0007919 pre-mRNA内含子的结合以及该pre-mRNA的剪接和环状化,从而生成hsa_circ_0007919。Hsa_circ_0007919通过以依赖LIG1的方式增强DNA损伤修复以维持细胞存活,从而促进GEM耐药。靶向hsa_circ_0007919和DNA损伤修复途径可能是PDAC的治疗策略。本文于2023年12月发表于“Molecular Cancer”(IF=37.3)上。
技术路线

结果:
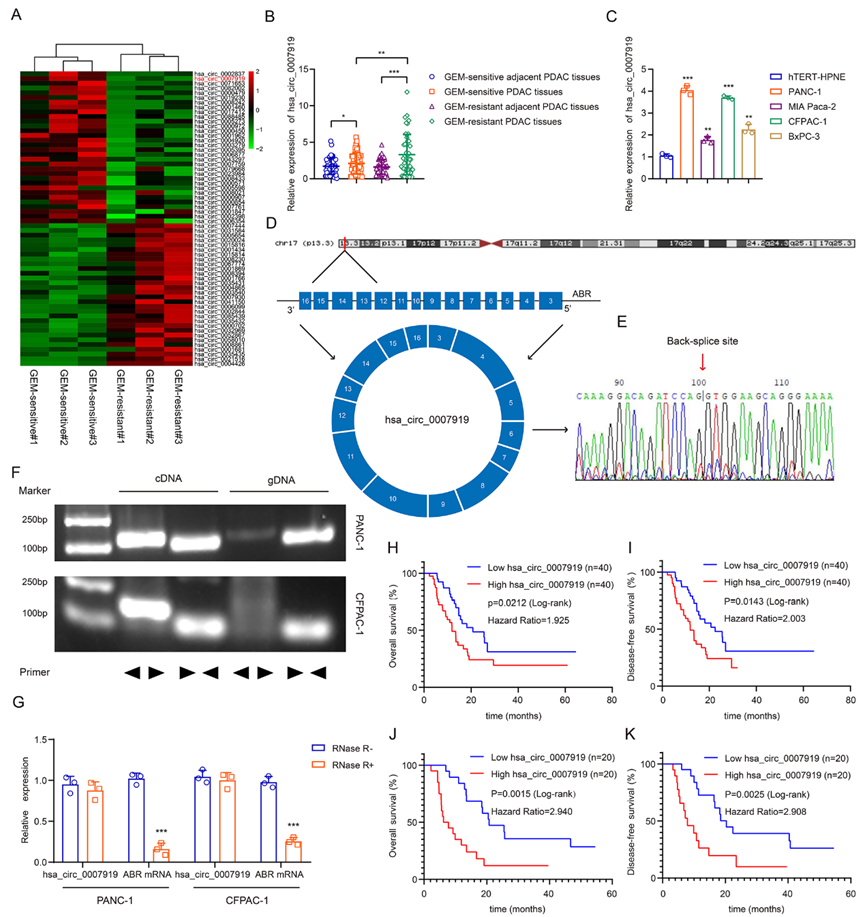
1)hsa_circ_0007919在GEM耐药PDAC中表达上调,预测预后不良
我们进行了下一代测序,以鉴定导致GEM耐药的环状RNA。共有62个circRNA差异表达,与GEM敏感组织相比,hsa_circ_0007919在GEM耐药PDAC组织中显著上调(图1A)。然后,我们测量了50对非GEM耐药PDAC组织及邻近组织和45对GEM耐药PDAC组织及相关邻近组织中hsa_circ_0007919的表达。结果显示,与GEM敏感的PDAC组织相比,hsa_circ_0007919在GEM耐药PDAC组织中的表达明显上调(图1B)。hsa_circ_0007919在PDAC细胞中表达增加,包括PANC-1、CFPAC-1、BxPC-3和MIA-Paca2,其表达水平在PANC-1和CFPAC-1细胞中相对较高(图1C)。接下来,我们评估了hsa_circ_0007919的结构,该结构来源于ABR基因的外显子3-16,并通过Sanger-seq验证了hsa_circ_0007919的环状位点(图1D-1E)。我们还设计了发散型和收敛型引物来检测hsa_circ_0007919在cDNA和gDNA中的表达。结果表明,hsa_circ_0007919可以从cDNA中扩增出来,但不能从gDNA中扩增出来(图1F), hsa_circ_0007919对RNase R外切酶酶切的抗性证实了它确实是环状的(图1G)。最后,我们利用临床组织样本数据分析hsa_circ_0007919表达与临床病理特征的相关性。我们分析hsa_circ_0007919表达与GEM耐药患者总生存期(OS)和无病生存期(DFS)的关系显示,hsa_circ_0007919高表达预示较差的OS和DFS (图1H-1I)。此外,我们将40例GEM治疗的PDAC患者分为hsa_circ_0007919高表达和低表达两组,发现hsa_circ_0007919高表达同样预测较差的OS和DFS(图1J-1K)。
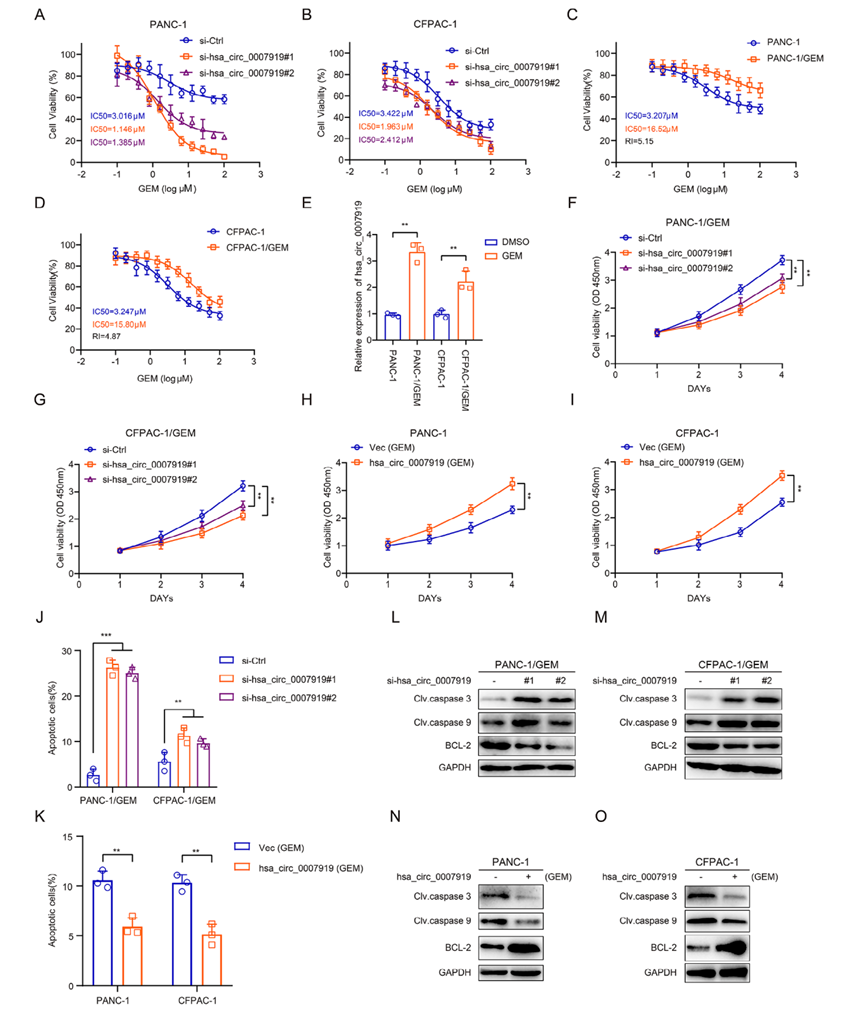
2)hsa_circ_0007919抑制DNA损伤和吉西他滨敏感性
由于hsa_circ_0007919在GEM耐药PDAC组织中表达上调,我们研究了它在GEM耐药细胞中的功能。首先,我们在PDAC细胞中沉默了hsa_circ_0007919,发现抑制hsa_circ_0007919增加了GEM的敏感性(图2A-2B)。然后,我们构建了GEM耐药PDAC细胞系PANC-1/GEM和CFPAC-1/GEM(图2C-2D),发现hsa_circ_0007919在这些GEM耐药细胞中高表达(图2E)。我们再次在这两种GEM耐药细胞系中沉默hsa_circ_0007919,并在GEM处理的正常PANC-1和CFPAC-1细胞中过表达hsa_circ_0007919 (图2C-2D)。CCK-8实验、FCM实验和DNA Ladder实验结果表明,沉默hsa_circ_0007919可降低细胞增殖,增加细胞凋亡,而过表达hsa_circ_0007919则相反(图2F-2K)。与凋亡实验结果一致,hsa_circ_0007919沉默增加了cleaved caspase 3和cleaved caspase 9的水平,降低了BCL2的表达,而hsa_circ_0007919过表达降低了cleaved caspase 3 and cleaved caspase 9的水平,增加了BCL2水平(图2L-2O)。GEM作为一种嘧啶类抗代谢药物,可诱导单基损伤并导致DNA断裂,因此我们评估了hsa_circ_0007919对DNA损伤的影响,发现hsa_circ_0007919沉默增加了单细胞凝胶电泳尾部和细胞核中γ-H2AX的积累,而hsa_circ_0007919过表达降低了这些参数(图3A, 3B)。最后,我们在裸鼠身上建立异种移植模型,发现沉默hsa_circ_0007919的PANC-1/GEM和PANC-1/GEM细胞形成的肿瘤体积和重量比对照细胞形成的肿瘤体积和重量都减少(图3C-3G)。IHC、TUNEL和qPCR结果显示,沉默hsa_circ_0007919可降低Ki-67的表达,增加caspase3和γ-H2AX的表达和细胞凋亡(图3H-3J)。这些结果表明,hsa_circ_0007919通过减少DNA损伤促进PDAC细胞增殖和减少细胞凋亡,从而增强PDAC细胞的GEM耐药性。
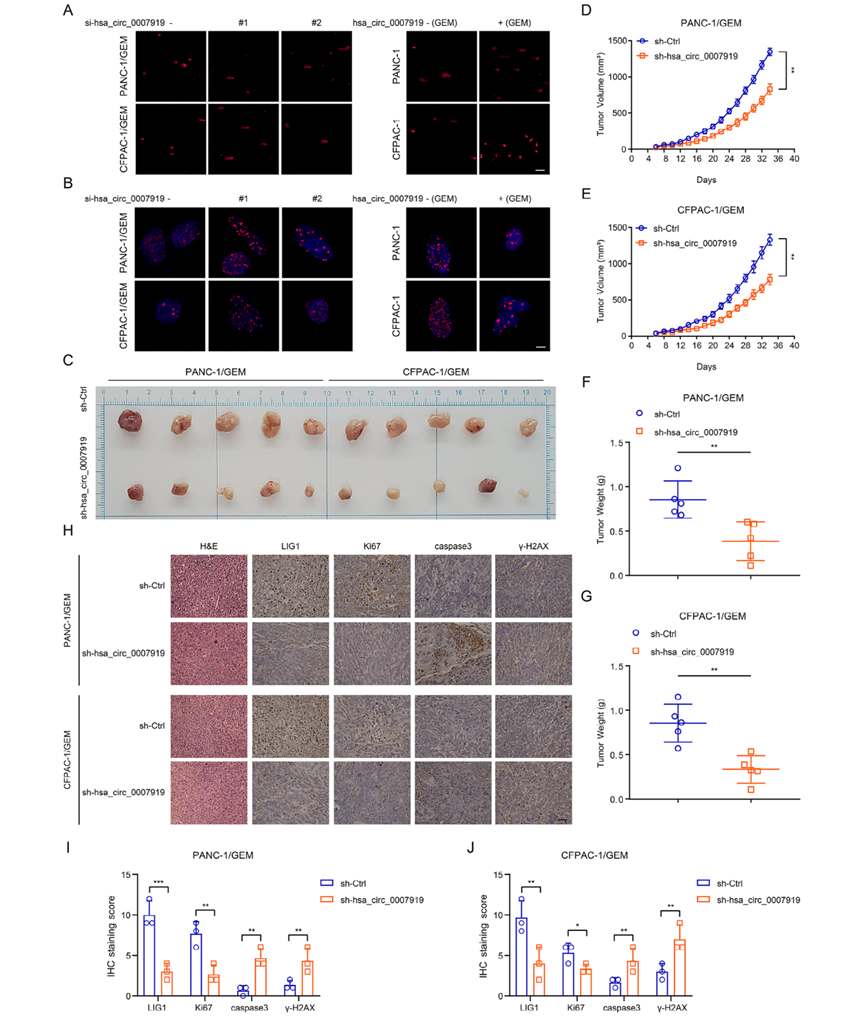
3)hsa_circ_0007919通过LIG1介导的修复途径抑制DNA损伤
为了证实hsa_circ_0007919如何抑制DNA损伤并影响GEM敏感性,我们通过RNA-seq鉴定了hsa_circ_0007919沉默的PANC-1/GEM细胞与对照细胞的差异表达基因。有520个上调基因和219个下调基因(图4A), KEGG分析和GSEA显示,这些基因在多种DNA损伤修复途径中富集,包括碱基切除修复、错配修复和核苷酸切除修复(图4B-4E)。LIG1是所有这些通路中共有的下调幅度最大的基因(图4F)。LIG1是DNA连接酶家族的一员,据报道,在几乎所有DNA损伤修复途径中,LIG1都在DNA重组中发挥重要作用。因此,我们测量了LIG1的表达,发现与正常PDAC组织相比,LIG1在GEM耐药PDAC组织中也高表达,并且与hsa_ circ_0007919在PDAC组织中的表达呈正相关(图4G–4I)。在hsa_circ_0007919沉默后,LIG1的mRNA和蛋白表达水平降低,而当hsa_circ_0007919过表达时,LIG1的mRNA和蛋白表达水平升高(图4J-4M)。这些结果表明,hsa_circ_0007919诱导LIG1表达激活DNA损伤修复途径,增强PDAC细胞对GEM的抗性。
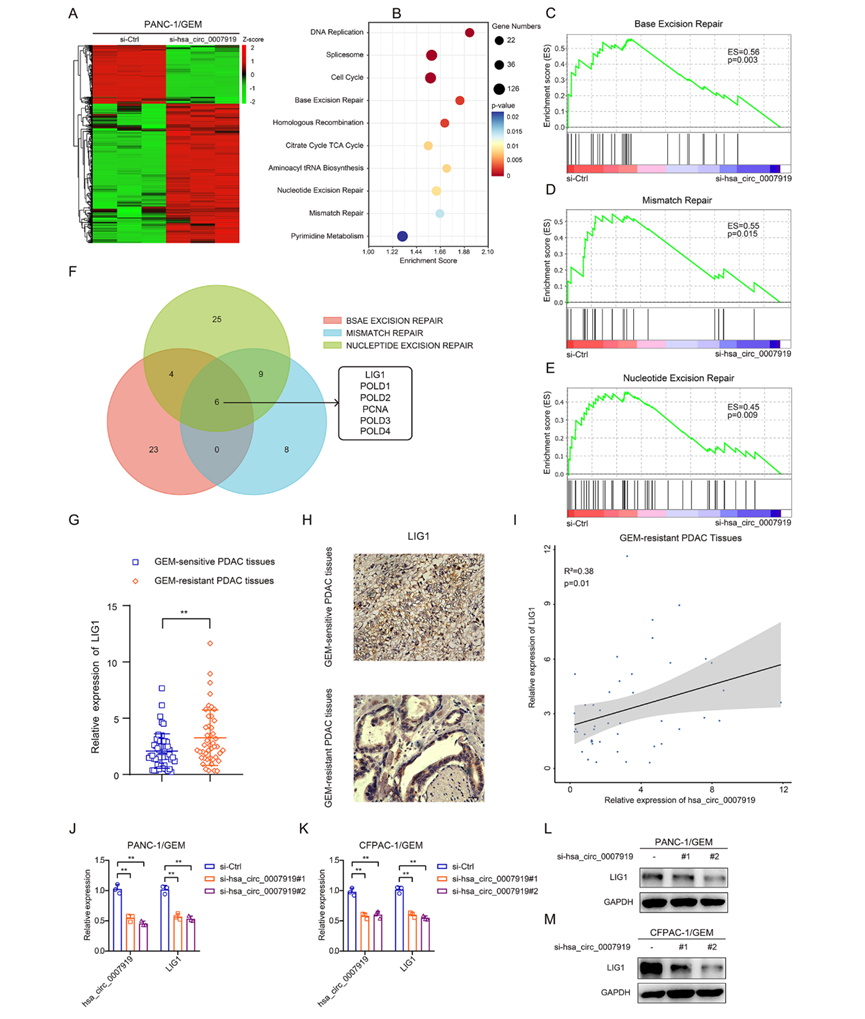
4)LIG1逆转了hsa_circ_0007919对细胞增殖、凋亡和DNA损伤的影响
为了证实LIG1是hsa_circ_0007919的下游靶点,我们研究了LIG1在GEM耐药PDAC细胞中的作用,发现沉默LIG1导致增殖减少,细胞凋亡和DNA损伤增加(图5A-5G)。此外,我们进一步在hsa_circ_0007919沉默稳定的PANC-1/GEM和PANC-1/GEM细胞中过表达LIG1,发现LIG1过表达逆转了hsa_circ_0007919沉默影响的细胞增殖、凋亡和DNA损伤(图5H-5N)。这些结果表明,hsa_circ_0007919通过增加LIG1的表达,促进细胞增殖,减少细胞凋亡和DNA损伤。
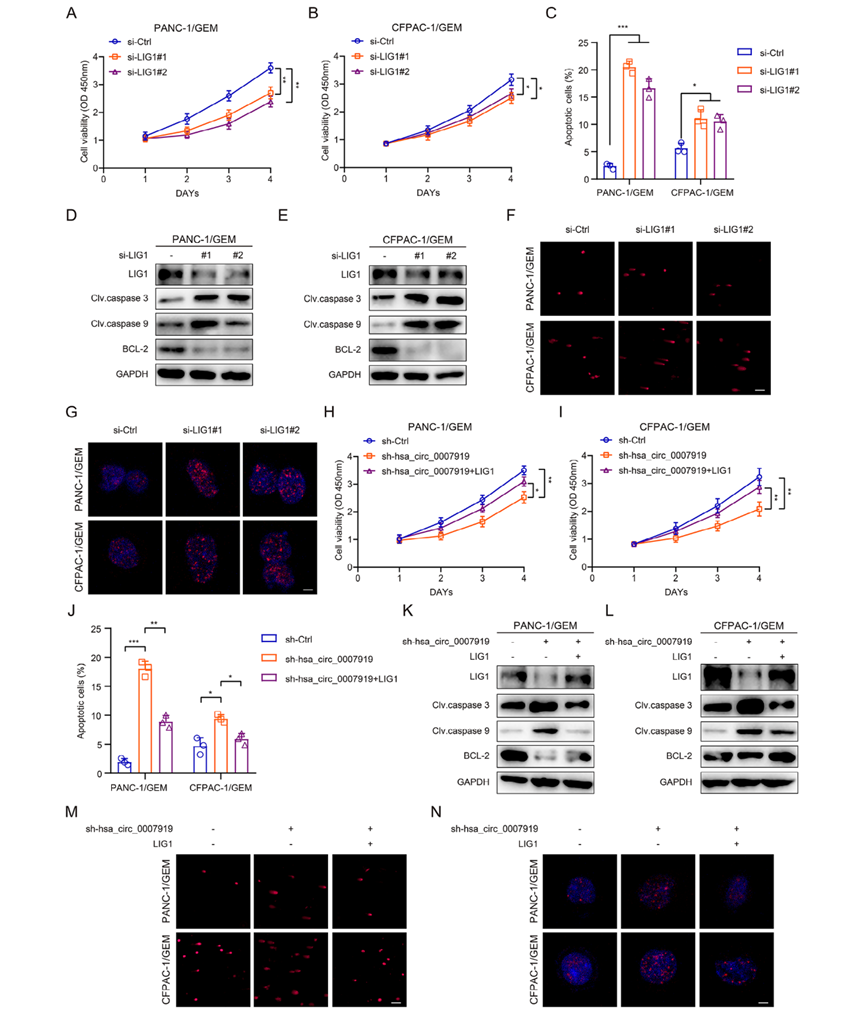
5)hsa_circ_0007919结合FOXA1和TET1促进LIG1转录
为了研究hsa_circ_0007919如何增加LIG1的表达,我们进行了FISH和核细胞质RNA分离实验,结果显示hsa_circ_0007919主要分布在细胞核中(图6A-6B)。我们使用circAtlas 2.0和ENCORI数据库确定了与hsa_circ_0007919结合的蛋白和与LIG1 mRNA结合的蛋白之间的重叠,但无法识别任何重叠蛋白。然后,我们使用circAtlas 2.0和SPP数据库确定了与hsa_circ_0007919结合的蛋白质和与LIG1启动子结合的蛋白质之间的重叠,FOXA1被确认为蛋白质最重要的重叠(图6C)。我们进一步预测,可能还有其他蛋白质与FOXA1一起起作用,并鉴定出与FOXA1结合的TET1 (图6D)。由于FOXA1在多种癌症中作为转录启动子,而TET1作为DNA甲基化酶降低各种基因启动子的甲基化水平并增强其转录,我们预测hsa_circ_0007919结合FOXA1和TET1促进LIG1的转录。我们首先沉默FOXA1和TET1,发现LIG1的表达降低(图6E-6H),co-IP实验的结果证实了FOXA1和TET1在GEM耐药细胞中的相互作用(图6I-6J)。同时,我们在FOXA1沉默的GEM耐药细胞中沉默TET1,发现TET1可以增强FOXA1沉默对LIG1的抑制能力,而过表达TET1可以部分逆转FOXA1沉默对LIG1的抑制能力,这表明FOXA1和TET1在调节LIG1中发挥协同作用(图6K-6L)。然后,我们进行了ChIRP分析,发现hsa_ circ_0007919可以结合FOXA1和TET1(图6M, 6N)。此外,我们使用RIP实验证实FOXA1和TET1可以与hsa_circ_0007919相互作用,且这种相互作用在GEM耐药细胞中得到增强(图6O-6P)。
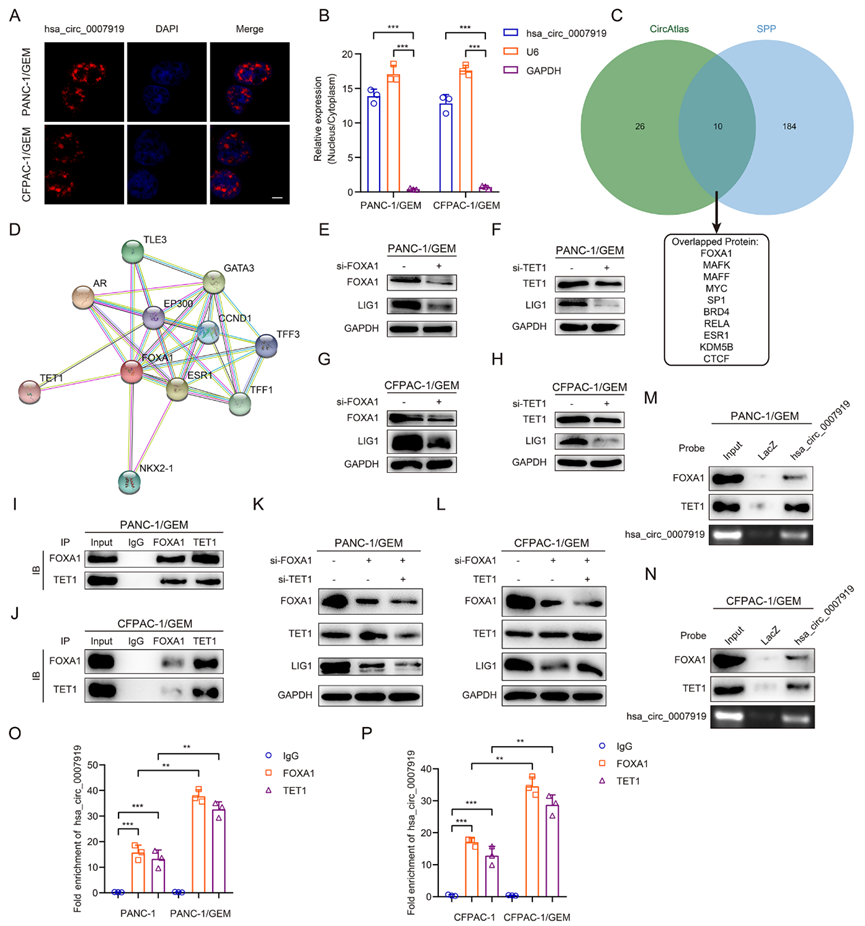
为了研究FOXA1、TET1与LIG1启动子之间的相互作用,我们使用JASPAR数据库分析了FOXA1与LIG1启动子之间的结合位点,并使用MethPrimer 2.0数据库预测了LIG1启动子中的CpG岛。在JASPAR鉴定的6个位点和4个预测的CpG岛中,我们发现LIG1启动子的位点6富集程度最高,因此我们选择了- 1411至-1273区域(P1)进行进一步研究(图7A-7B),我们发现5-AzaC处理增加了GEM耐药细胞中LIG1的表达(图7C)。ChIP实验结果显示FOXA1和TET1结合到LIG1启动子区域P1。抑制hsa_circ_0007919降低了这种结合能力(图7D)。MS-PCR检测结果显示,沉默hsa_circ_0007919或TET1会增加LIG1启动子中的DNA甲基化水平(图7F),而过表达hsa_circ_0007919或TET1具有相反的效果(图7E和7G)。此外,我们建立了一个荧光素酶报告基因实验,发现沉默hsa_circ_0007919、FOXA1或TET1会降低LIG1启动子的转录活性,而过表达hsa_circ_0007919、FOXA1或TET1会增强LIG1的转录活性(图7H -7I)。这些结果表明hsa_circ_0007919通过结合FOXA1和TET1增强了LIG1的转录。
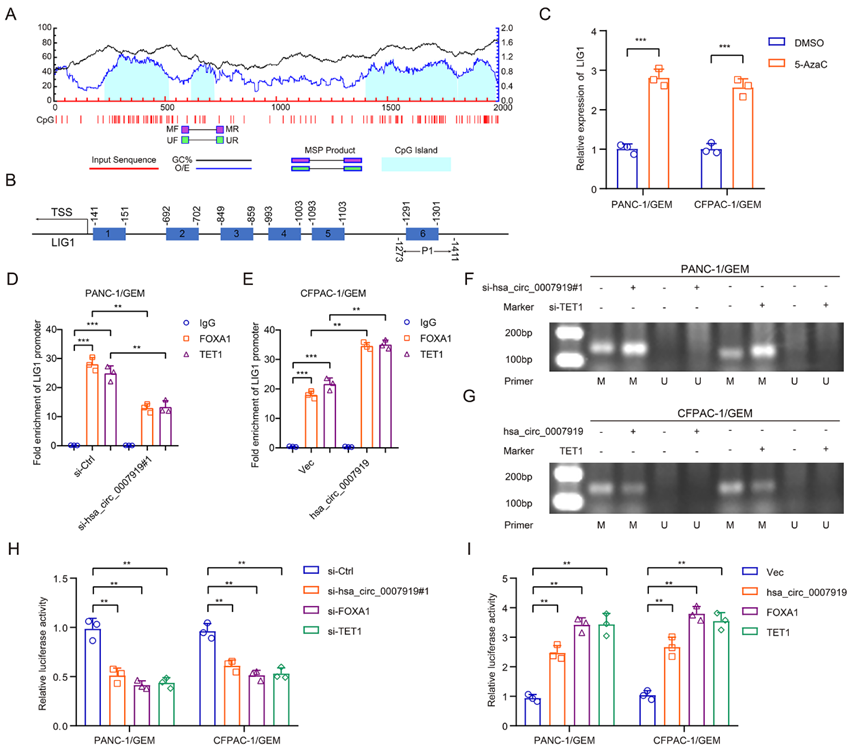
6)吉西他滨通过增强QKI介导的反向剪接诱导hsa_circ_0007919表达
研究表明,在circRNA合成过程中,有多种蛋白质参与了反向剪接过程。在几个公认的调控因子中,有报道称QKI和FUS可以促进环状RNA的形成,而ADAR1则起到相反的作用。为了探究hsa_circ_0007919高表达的原因,我们分析了上述蛋白与hsa_circ_0007919在耐药PDAC组织中的表达相关性,发现QKI与hsa_circ_0007919表达呈正相关,FUS与hsa_circ_0007919表达相关性较低,ADAR1与hsa_circ_0007919表达呈负相关(图8A)。因此,我们预测QKI可以促进hsa_circ_0007919的形成。我们在GEM耐药PDAC细胞中沉默QKI,发现hsa_circ_0007919的表达下调,但hsa_circ_0007919宿主基因ABR的表达不受影响(图8B-8C);此外,QKI的表达在正常PDAC细胞和抗GEM PDAC细胞之间没有差异(图8D-E8)。据报道,QKI在其pre-mRNA中与环状RNA外显子侧翼的内含子相互作用。我们设计了ABR内含子2和16的引物,发现QKI可以在PDAC细胞中与这两个内含子结合,并且这种相互作用在耐GEM的PDAC细胞中得到增强(图8F-8G)。综上所述,本研究表明GEM增强QKI介导的hsa_circ_0007919剪接和循环化,以及hsa_circ_0007919招募FOXA1和TET1来调节LIG1转录和DNA损伤修复途径,从而有助于抵抗GEM诱导的PDAC细胞DNA损伤和凋亡(图8H)。
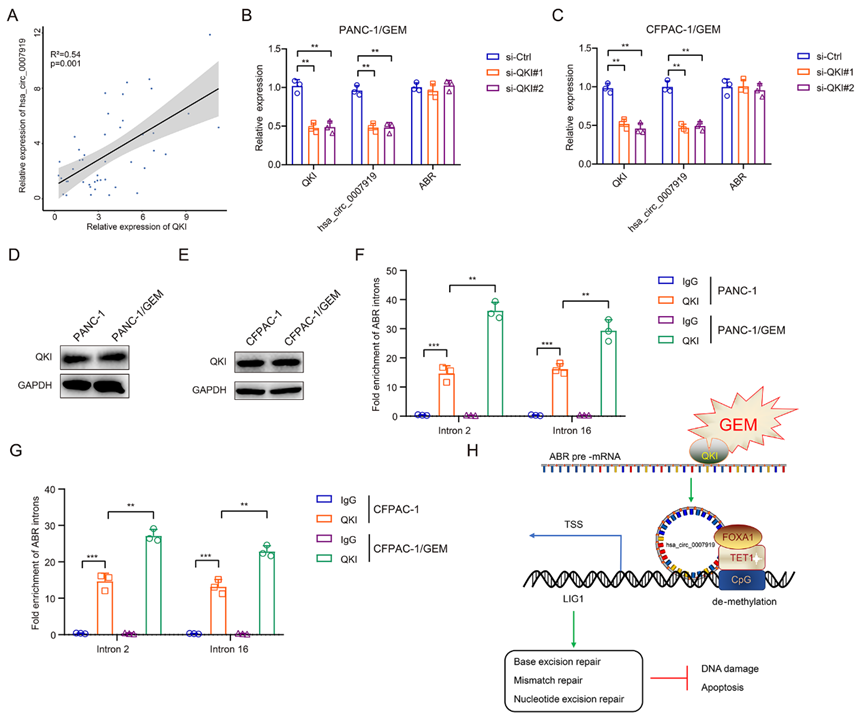
结论:我们的研究结果表明hsa_circ_0007919可以促进DNA损伤修复以对抗GEM治疗。机制上,hsa_circ_0007919招募FOXA1和TET1促进LIG1转录,激活碱基切除修复、错配修复和核苷酸切除修复途径,改善DNA损伤,抑制GEM诱导的细胞凋亡。此外,GEM处理增强了QKI和ABR pre-mRNA之间的相互作用,以反向剪接依赖的方式导致hsa_circ_0007919增加生物发生。我们的发现可能有助于了解GEM耐药的机制,并制定化疗耐药PDAC的治疗策略。
实验方法:qRT-PCR,CCK-8,流式,WB,免疫荧光,IHC,动物实验,TUNEL,GSEA,FISH,ChIRP,Co-IP,RIP,ChIP,MS-PCR,荧光素酶报告试验。
参考文献:Xu L, Ma X, Zhang X, Zhang C, Zhang Y, Gong S, Wu N, Zhang P, Feng X, Guo J, Zhao M, Ren Z, Zhang P. hsa_circ_0007919 induces LIG1 transcription by binding to FOXA1/TET1 to enhance the DNA damage response and promote gemcitabine resistance in pancreatic ductal adenocarcinoma. Mol Cancer. 2023 Dec 4;22(1):195. doi: 10.1186/s12943-023-01887-8.