






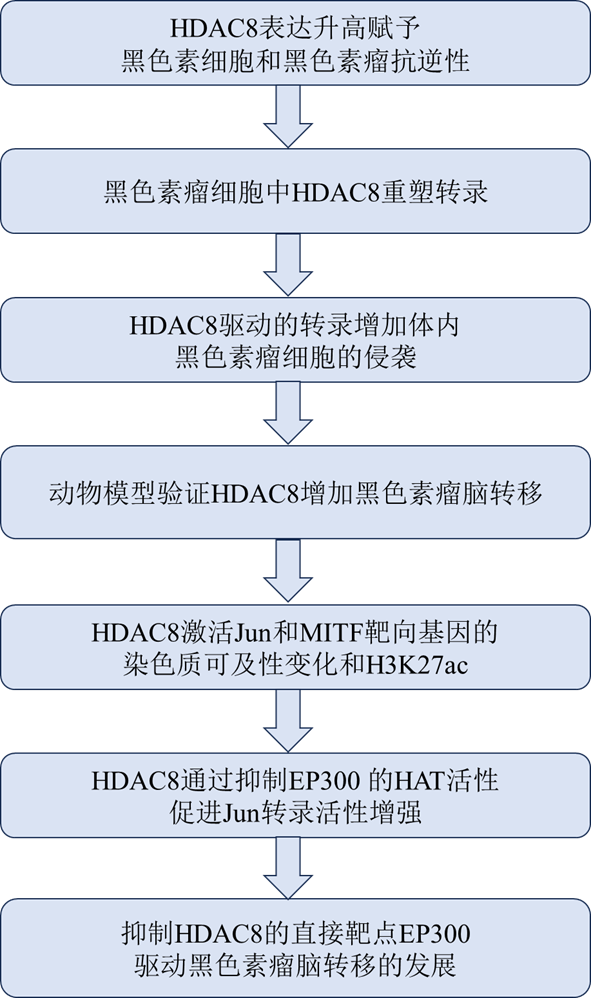
黑色素瘤脑转移信号通路:HDAC8介导EP300 抑制驱动转录状态
皮肤黑色素瘤是最致命的皮肤癌类型,有向多个器官转移的倾向。黑色素瘤转移发展的一个常见部位是大脑,如果不治疗,黑色素瘤脑转移(Melanoma Brain Metastases,MBMs)迅速进展,大多数患者在3个月内死亡。对MBM发展的分子驱动因素知之甚少。迄今为止,大多数研究都集中在PTEN丢失和AKT信号过度激活上,这限制了对于黑色素瘤异质性的调节。本研究确定应激诱导的HDAC8活性驱动了黑色素瘤脑转移发展,通过ATAC-Seq和ChIP-Seq表明HDAC8活性增加是通过H3K27ac和c-Jun结合位点可及性的增强改变了染色质结构。HDAC8被鉴定为转录辅因子失活和染色质可及性的介质,驱动脑转移。
该研究于2023年11月29日发表在《Nature Communications》,IF:16.6
技术路线
主要研究结果
1. 黑色素瘤细胞受到压力后HDAC8活性增加
作者首先确定了HDAC8是否是黑色素瘤细胞状态变化的表观遗传驱动因素。黑色素瘤细胞的WB分析表明,黑色素瘤细胞谱系转录因子MITF的高表达与较低的HDAC8表达相关,并降低由HDAC8调节的信号分子水平,包括SMC3乙酰化、EGFR磷酸化和c-Jun磷酸化(图1a)。根据细胞对BRAF抑制剂(BRAFi)vemurafenib的基线敏感性对其进行排名,并证明高MITF表达、低HDAC8表达和高BRAFi敏感性之间的联系(图1a)。同样, HDAC8低表达的黑色素瘤细胞中过表达HDAC8后磷酸化Jun和EGFR水平增加(图1b)。由于转录转换可能是对外源性应激的反应,作者接下来探讨了HDAC8的过表达是否会增加不同微环境条件下黑色素细胞的存活。HDAC8表达的增加导致暴露于缺氧、紫外线照射和BRAF-MEKi治疗后的黑色素瘤细胞存活增加(图1c-e)。为了确定这是否与黑色素细胞和黑色素瘤细胞间的保守反应有关,用紫外线照射处理了2个原代人类黑色素细胞系(FOM173和NHEM),并发现HDAC8和磷酸化c-Jun表达增加(图1f)。人类黑色素细胞系(HERMES1和HERMES3)中HDAC8表达升高导致MITF表达下降,磷酸化c-Jun水平增加(图1g),并在细胞受到缺氧或紫外线照射时提高了存活率(图1h&i)。
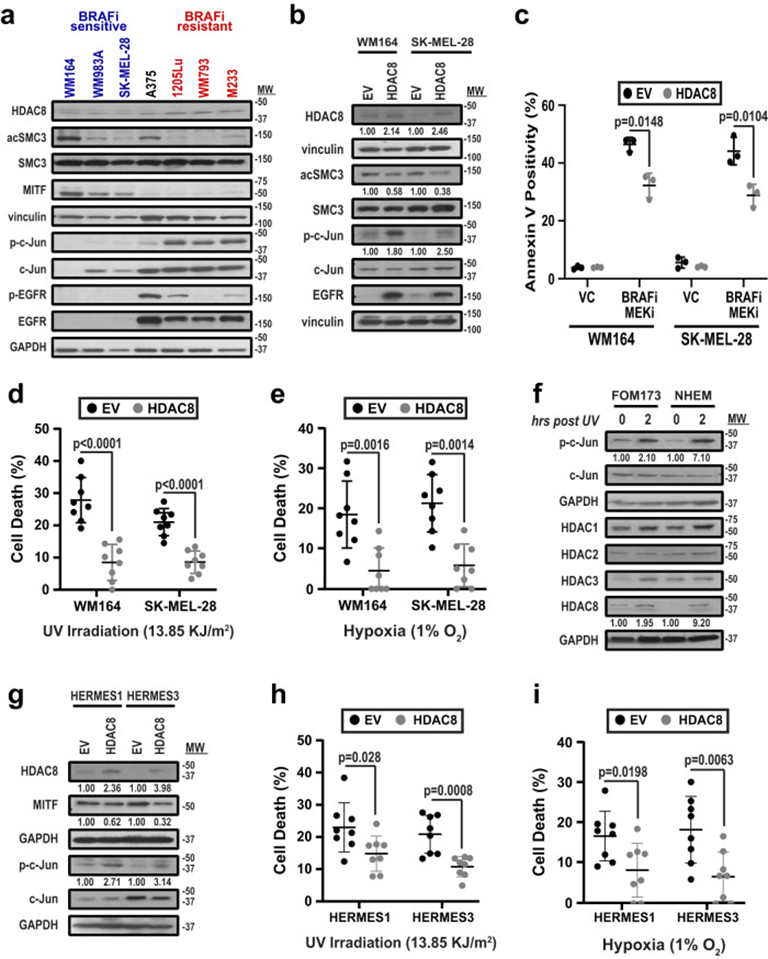
图1:HDAC8表达赋予黑色素细胞和黑色素瘤抗逆性
2. HDAC8驱动黑色素瘤细胞中的转录开关
对两个黑色素瘤细胞系(WM164和SK-MEL-28)进行RNA-seq分析,以确定HDAC8的上调如何重塑转录格局,发现随着HDAC8的表达升高,204个常见基因显著增加,163个常见基因显著减少(图2a)。KEGG分析显示,HDAC8高表达导致了黑色素瘤转移等途径的富集,包括PI3K-AKT信号、局灶粘附、ECM-受体相互作用、肌动蛋白细胞骨架调节、MAPK信号和RAP1信号(图2b),同时减少了一些代谢途径如谷胱甘肽和嘌呤代谢的富集(图2c)。这些数据表明HDAC8驱动的转录程序与侵袭性、转移性表型相关,接下来确定HDAC8表达增加是否与黑色素瘤转录状态相关。最近对黑色素瘤异质性的分析确定了几种不同的细胞状态,包括未分化胚胎干细胞(ESC)状态、神经嵴干细胞(NCSC)样状态和分化黑色素细胞状态等。对数据集的分析表明,HDAC8高表达富集了 WM164细胞中的NCSC和未分化状态相关的基因(图2d&e)。通过分析WM164和1205Lu细胞中NCSC基因启动子区域的H3K27ac 的ChIP-Seq数据,验证了NCSC基因转录活性的增加。与NCSC状态相关的基因,包括AXL、FAM83G、FOSL1和SOX2,增加了HDAC8过表达细胞中的H3K27ac乙酰化(图2f);而与黑色素细胞状态相关的基因,包括MLANA、DCT、TYR和PMEL,减少了HDAC8过表达细胞中的H3K27ac乙酰化(图2g)。
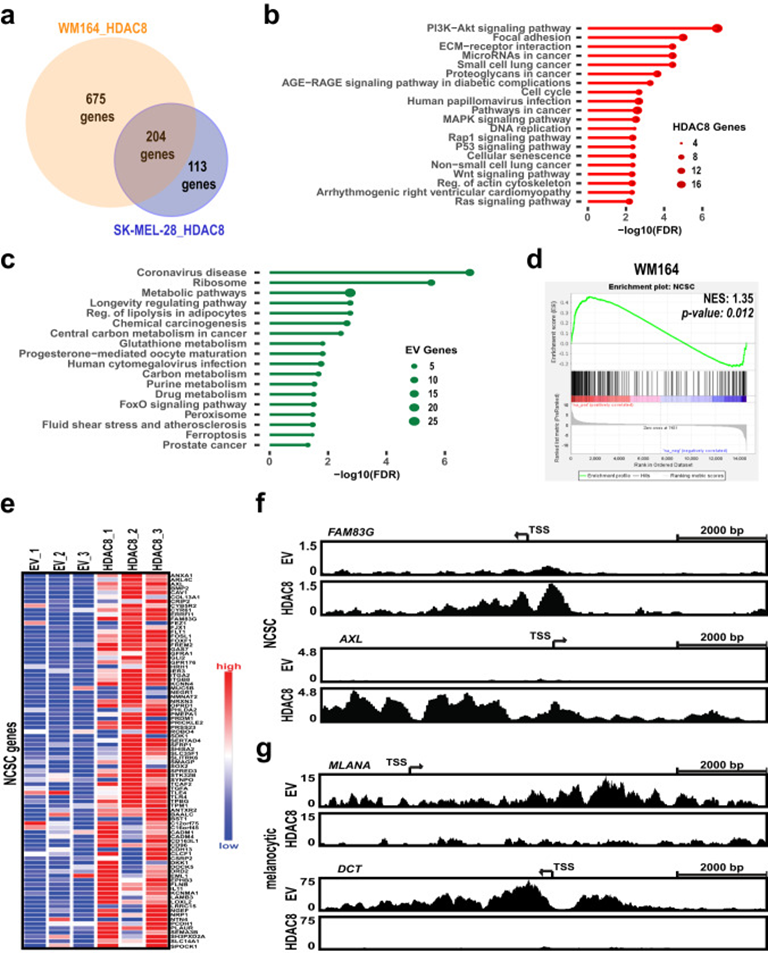
图2: HDAC8驱动黑色素瘤细胞中的转录开关
3. HDAC8驱动的转录状态导致侵袭性变形虫表型
转移到大脑的细胞必须克服多种障碍,包括通过血脑屏障外渗、在循环系统的高剪应力条件和脑实质富含蛋白酶的环境中生长。由于在RNA-seq和ChIP-Seq分析中上调的许多基因与侵袭增加有关,所以进行了3D球形胶原蛋白侵袭和基质胶侵袭检测,发现WM164和SK-MEL-28细胞在HDAC8过表达后细胞侵袭增强(图3a-d)。接下来观察HDAC8是否通过使用模拟一般循环中经历的剪切水平的流动室系统来增加黑色素瘤细胞对剪切应力的弹性。WM164和SK-MEL-28细胞暴露于高水平的流体剪切应力24h后,对照细胞高水平死亡(图3e&f)。HDAC8上调的许多基因参与细胞骨架重排,包括与变形虫表型相关的SOX2和PROM1。WM164和SK-MEL-28细胞表达HDAC8后被镀在胶原蛋白上时采用了圆形变形虫表型(图3g)。由于变形虫表型可以允许细胞挤过狭窄的空间,例如离开血管和穿过血脑屏障相关的空间,进行跨内皮细胞迁移检测,发现HDAC8过表达导致黑色素瘤细胞通过汇合内皮细胞层,转移性增加了十倍(图3h&i)。总的来说,这些发现表明HDAC8驱动的转录程序可以增加体内黑色素瘤细胞的侵袭。
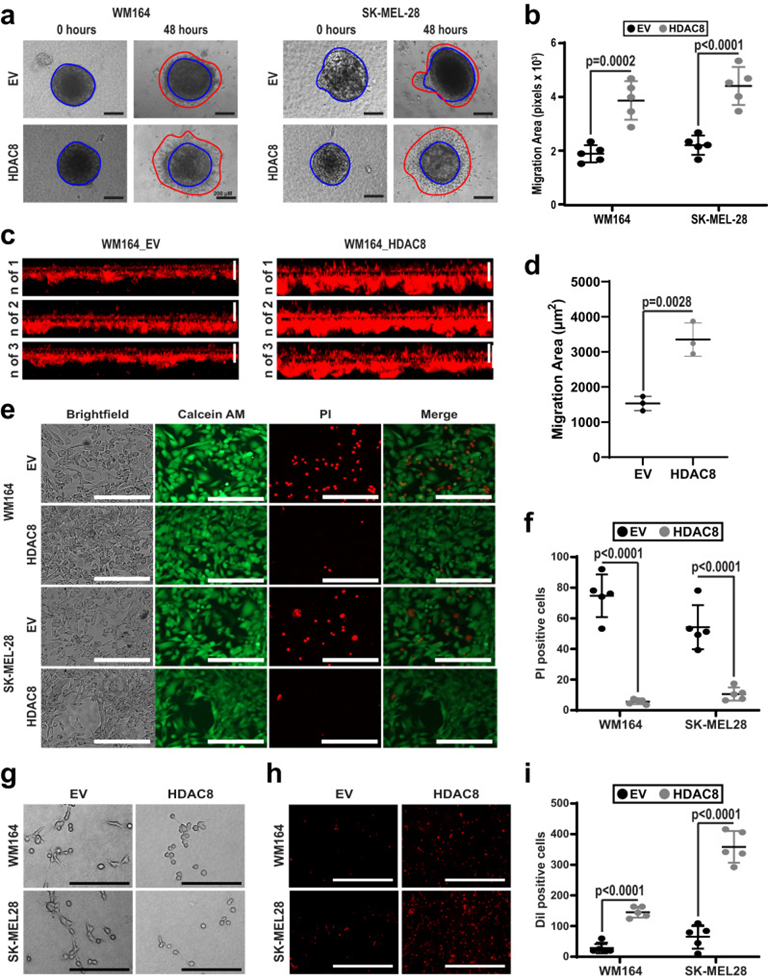
图3: HDAC8增加黑色素瘤细胞的侵袭和变形虫样状态
4. HDAC8增加小鼠脑转移的发展
接下来,作者检测HDAC8驱动的转录状态是否改变了体内转移播种的模式。对照组或表达HDAC8的WM164被注射到小鼠心脏左心室和每隔5天收集的器官中,以确定潜在的转移位点。过表达HDAC8和EV组之间肝脏或肺转移的总发生率差异不大(图4a)。与第5天的EV细胞相比,HDAC8表达后肝转移数量显着增加,但在之后的时间点不再显着(图4b)。对肺的分析显示,随着时间的推移,HDAC8表达组和EV对照组之间的转移发展动态相似(图4c)。作者接下来关注高表达HDAC8的黑色素瘤细胞形成黑色素瘤脑转移的倾向。在心内注射后, SK-MEL-28、WM164和1205Lu的高表达HDAC8黑色素瘤细胞与对照相比,出现更多的脑转移(图4d-f)。神经病理学检查确定肿瘤累及侧脑室颞角的室管膜表面,已经侵犯到脑实质,肿瘤边界侵犯血管周围。为了解决发现的临床相关性,作者对临床黑色素瘤样本进行单细胞RNA-seq分析。这些分析显示,人类黑色素瘤细胞中HDAC8表达与NCSC样基因表达谱之间存在显著相关性(图4g&h)。根据转移部位进行的更详细的细分显示,LMD和脑转移样本中HDAC8表达与NCSC样状态之间存在显著相关性,但与皮肤转移样本无关。然而,这可能是由于皮肤转移样本中肿瘤细胞数量较少。黑色素瘤脑转移通常富含OXPHOS代谢基因。
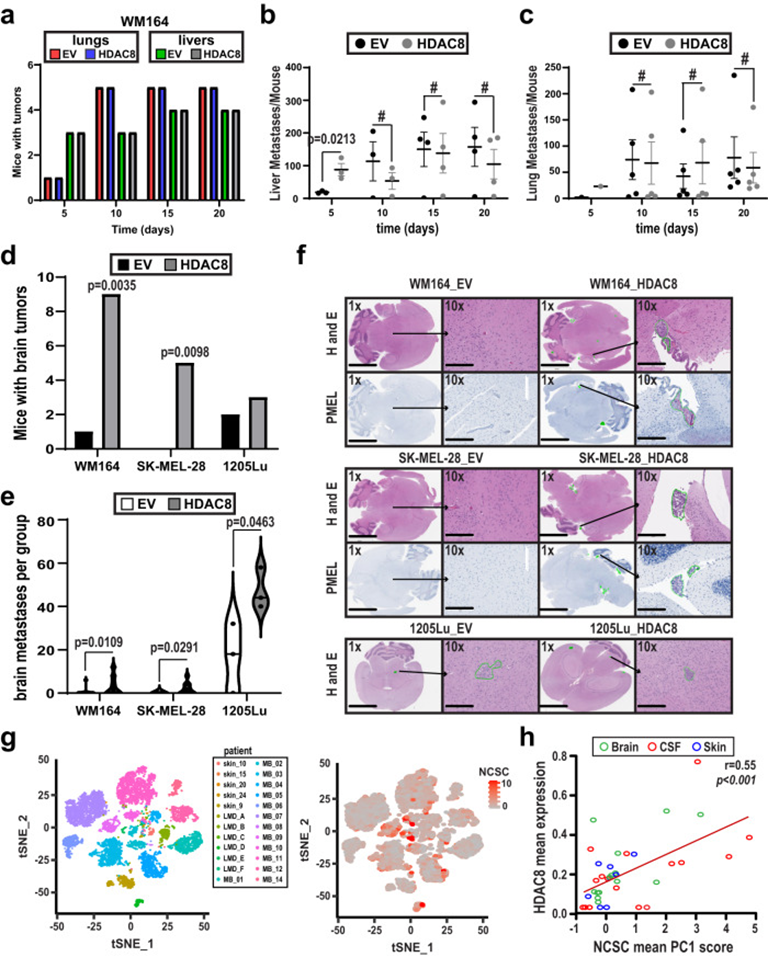
图4: HDAC8增加黑色素瘤脑转移的建立
5. HDAC8增加了Jun靶向基因的染色质可及性和H3K27ac
作者接下来使用ATAC-Seq和ChIP-Seq来确定HDAC8如何调节转录重编程。ChIP-Seq数据的整体分析显示HDAC8表达对H3K27ac在启动子或增强子区域几乎没有整体影响(图5a&b),总乙酰-H3组蛋白水平也几乎没有变化(图5c)。H3K27ac ChIP-Seq数据的HOMER分析显示,过表达HDAC8时,Jun位点的H3K27乙酰化增加,而对照细胞在对细胞周期进展重要的转录因子(包括B-myb和A-myb)上表现出H3K27乙酰化增加(图5d)。基因峰数/长度的ATAC-seq分析显示,在HDAC8过表达和EV细胞中,DNA片段大小相似(图5e)。虽然整个基因组的总体可及性相似,但在HDAC8表达细胞中,启动子的可及性增加(图5f),而在非启动子位点几乎没有差异(图5g)。对HDAC8表达时可及性增加的基序的分析包括多个AP-1转录因子(Fra1,ATF3,Jun)和TEADs(图5h,补充图10a,b)。多个基序在HDAC8引入时也显示下调,包括CTCF、多个SOX转录因子和黑色素细胞谱系转录因子MITF(图5h)。
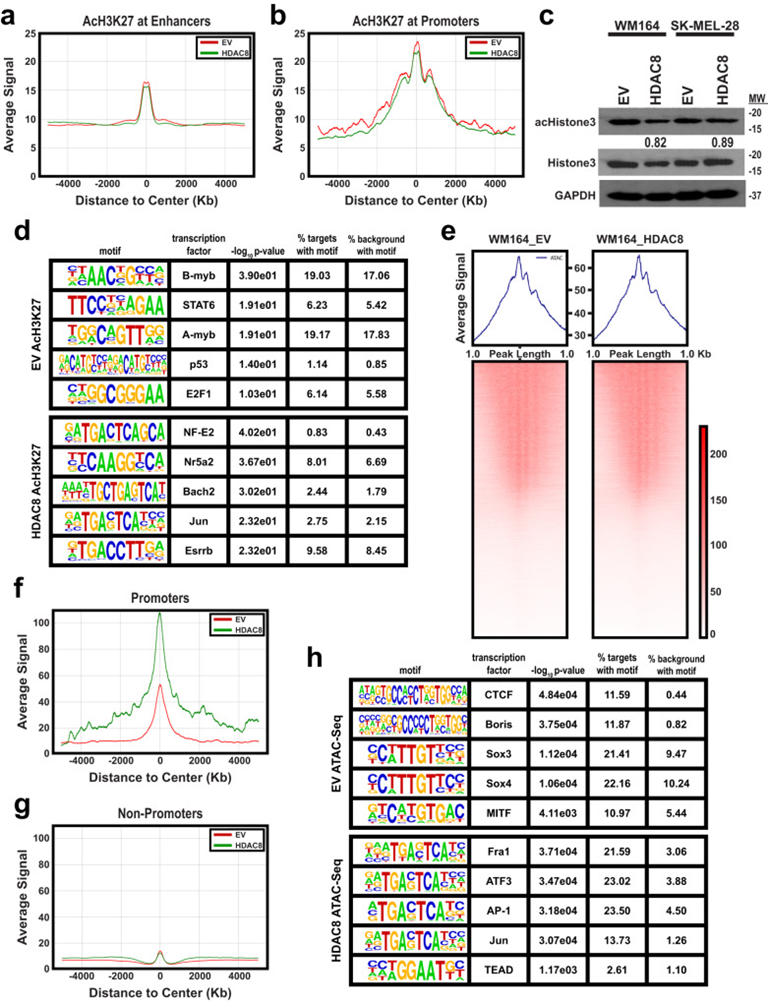
图5:HDAC8激活诱导Jun和MITF靶向基因的染色质可及性的变化
6. EP300失活导致Jun转录活性增加
ATAC-Seq和ChIP-Seq数据的生物信息学整合表明HDAC8表达增强细胞外基质组装、细胞分化相关的基因通路中的可及性/转录激活(图6a)。尽管ATAC-Seq和ChIP-Seq数据显示HDAC8介导的H3K27ac或染色质可及性的总体变化很少,但在WM164和1205Lu细胞系中都注意到离散Jun和MITF位点的变化。许多与NCSC表型有关的基因,如SOX2,由c-Jun调节,并在HDAC8过表达细胞中表现出可及性/H3K27乙酰化增加(图6b),而MITF调节的基因,如MLANA的可及性降低(图6c)。作者注意到HDAC8的表达导致多个Serpins(如Serpin E1,E2和A1)的基因表达,H3K27ac和染色质可及性增加,这些蛋白酶抑制剂有助于脑转移的建立。这些结果表明HDAC8的过表达导致从MITF驱动的转录程序到c-Jun调节的转录程序的转变。
由于HDAC8对全局染色质可及性的影响相对较小,接下来探究了HDAC8是否通过调节非组蛋白靶点发挥作用。进行蛋白质组学分析以确定HDAC8如何影响黑色素瘤细胞的“乙酰化”。 STRING分析发现参与脂质代谢调节的显著去乙酰化蛋白质(图6d),确定的中心枢纽之一是组蛋白乙酰转移酶(HAT)EP300/CREBBP。已知EP300可以乙酰化多种蛋白质,包括MITF和c-Jun。 EP300结构分析确定了它的HAT结构域中的4个赖氨酸残基(赖氨酸的1542、1546、1549和1550),它们在HDAC8过表达时被脱乙酰化(图6e)。HDAC8对EP300的脱乙酰化能力通过免疫沉淀和总蛋白乙酰化的免疫印迹(图6f)得到证实。在两个独立的黑色素瘤细胞系中,EP300的脱乙酰化与其HAT活性的降低(通过组蛋白H4的乙酰化测量)相关(图6g)。NCSC和黑色素细胞表型相关基因的单位点ChIP检测显示,HDAC8表达的增加与EP300与AXL的Jun启动子的结合增加以及EP300与MLANA的MITF启动子的结合减少有关(图6h)。这些结果表明HDAC8通过抑制其HAT活性将其从MITF启动子位点转换为Jun启动子位点来调节EP300功能。
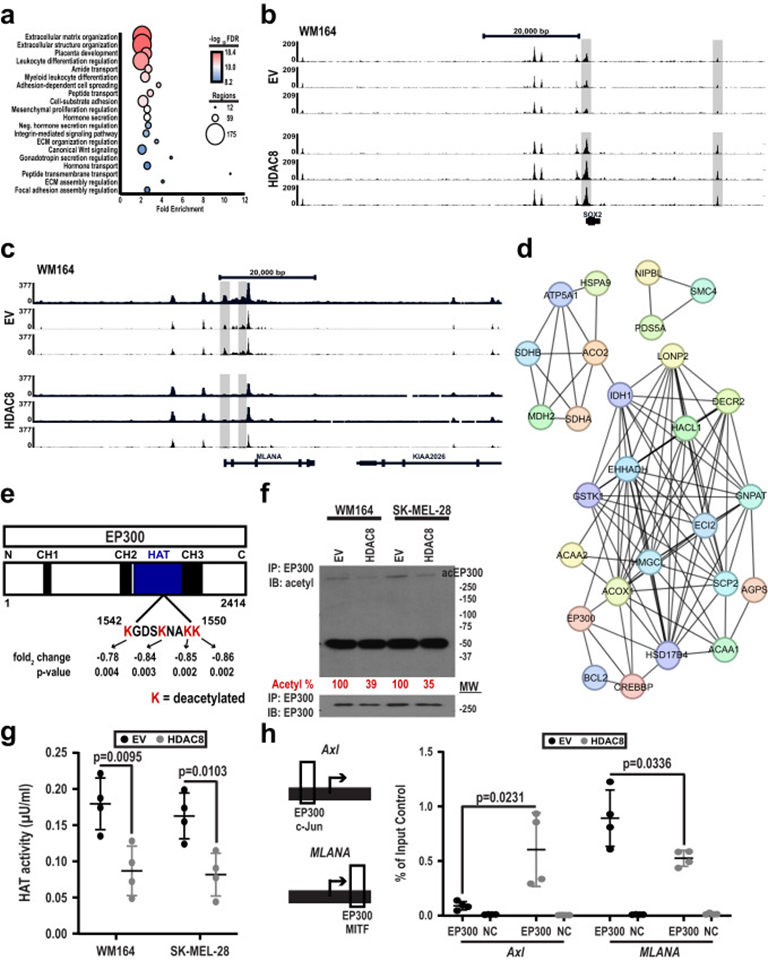
图6: HDAC8去乙酰化失活EP300,导致JUN转录活性增加
7. 抑制EP300驱动黑色素瘤脑转移的发展
由于EP300是HDAC8的直接靶点,作者接下来探索了EP300在驱动抗应激、侵袭表型中的作用。首先确定了对BRAFi治疗敏感或耐药的黑色素瘤细胞系中EP300和CREBBP的表达水平,并发现对抑制剂治疗敏感的细胞系EP300水平增加(图7a)。对TCGA黑色素瘤数据库的研究强调了与EP300/HDAC8水平不变的患者相比, EP300水平降低和HDAC8水平升高与总体生存率降低之间的相关性。然后进行功能研究以确定抑制EP300(通过HDAC8模仿其去乙酰化)是否会改变黑色素瘤细胞对BRAFi治疗的敏感性。BRAF突变黑色素瘤细胞系与EP300/CREBBP抑制剂I-CBP112联合治疗降低了黑色素瘤细胞对BRAFi的敏感性,增加了形成的菌落数量(图7b&c)。通过特异性siRNA沉默EP300也有类似的效果,并被发现BRAFI-MEKi治疗诱导的细胞凋亡水平降低(图7d)。这种影响与c-Jun活性的增加有关,发现抑制EP300可以增加转录活性的磷酸化c-Jun水平(图7e)。进一步的研究表明,通过shRNA敲低沉默EP300也保护WM164和SK-MEL-28黑色素瘤细胞免受紫外线照射和缺氧损伤(图7f&g)。由于表达HDAC8的细胞具有高度侵袭性,接下来研究调节EP300的表达是否会改变黑色素瘤细胞的侵袭能力。通过shRNA敲除沉默EP300增加了黑色素瘤细胞对3D胶原基质的侵袭(图7h-j)。因此,与shRNA对照组相比,心内注射EP300沉默的SK-MEL-28细胞增加了脑转移小鼠的数量(图7k&l)。综上表明HDAC8介导的EP300去乙酰化导致HAT活性降低,转录活性JUN增加,以及从黑色素细胞转换为NCSC样。
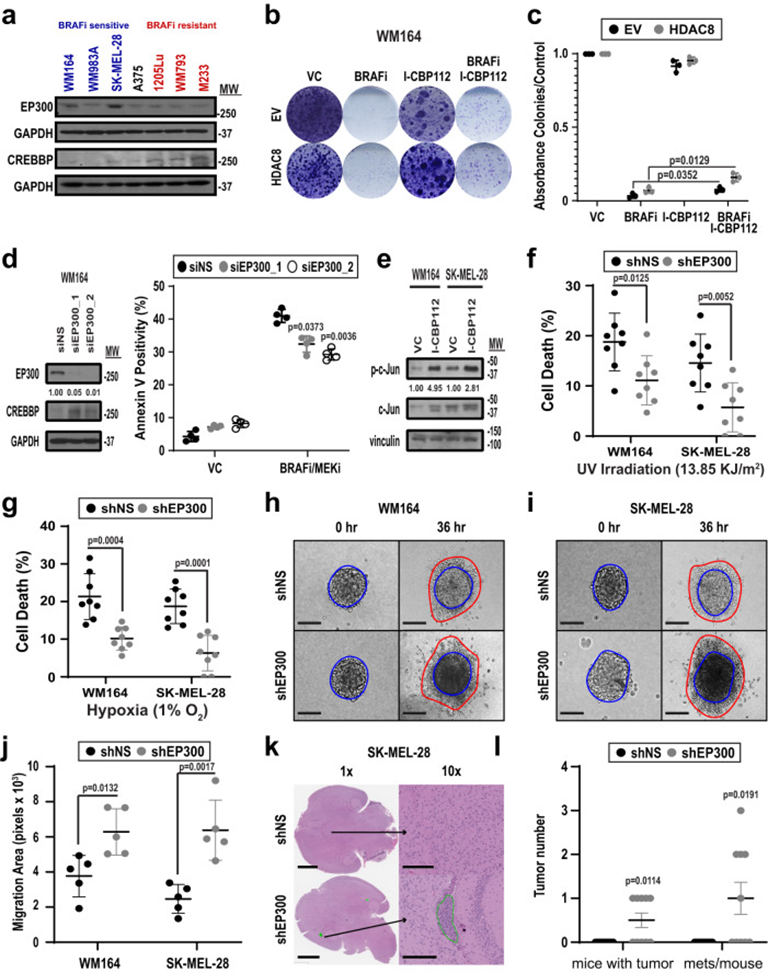
图7: EP300的抑制作用驱动了黑色素瘤细胞中的抗压侵袭性表型
结论
本研究证明在黑色素瘤细胞中,HDAC8介导的去乙酰化导致从MITF到Jun驱动的转录程序的转换,从而导致采用NCSC状态特征的转录状态。Jun蛋白的去乙酰化增加了其磷酸化和转录活性,导致Jun/AP-1驱动类似NCSC的转录程序的启动。与此同时,HDAC8介导的EP300去乙酰化抑制其HAT功能,降低MITF启动子位点的可及性,并改变H3K27在关键黑色素细胞基因中的分布。HDAC8诱导的转录程序导致黑色素瘤细胞采用高度弹性的变形虫表型,从而在剪切应力条件下和脑实质中驱动侵袭和存活。这种HDAC8驱动的程序也在暴露于紫外线照射的黑色素细胞中观察到,提供了保守的黑色素细胞存活程序和黑色素瘤细胞侵袭性转移行为之间的联系。
实验方法
细胞培养,质粒的生成和转染,免疫印迹,免疫沉淀,细胞活力测定,附着力/迁移分析,HAT活性测定,RNA测序和数据分析,ChIP测序和数据分析,ATAC测序和数据分析,scRNA-Seq数据分析,乙酰组学,小鼠体内试验,HE染色,免疫组织化学分析
参考文献
Emmons Michael F, Bennett Richard L, Riva Alberto et al. HDAC8-mediated inhibition of EP300 drives a transcriptional state that increases melanoma brain metastasis. [J] .Nat Commun, 2023, 14: 7759.