






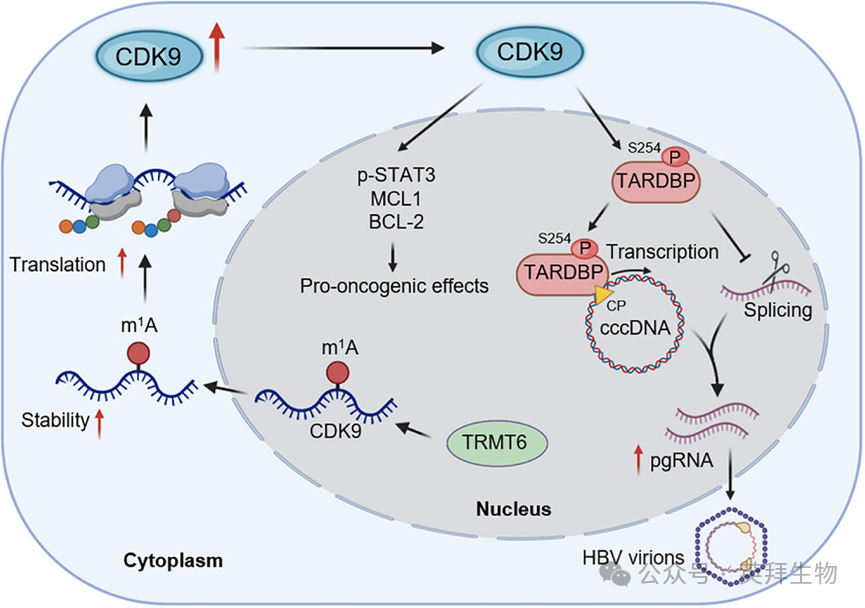
TRMT6介导的CDK9 mRNA的m1A修饰是HBV相关肝细胞癌的双重致病驱动因子
肝细胞癌(HCC)是全球范围内导致死亡的主要原因之一,其中乙型肝炎病毒(HBV)感染是主要的危险因素。mRNA甲基化的失调参与了肿瘤发生和病毒复制过程。然而,N1-甲基腺嘌呤(m1A)修饰与HCC进展及HBV复制之间的关联尚不明确。本研究通过对4例HCC组织和7例癌旁组织(其中2例来自本研究,5例来自GSE242889数据集)进行单细胞核RNA测序(snRNA-seq),发现HCC组织中mRNA甲基化水平升高,其中m1A“写入器”和“读取器”的表达增加,而m1A“擦除器”的表达降低。在这些分子中,m1A写入器TRMT6在HCC中表达上调,且与患者不良预后相关。敲低TRMT6显著抑制了HCC细胞的恶性表型、致瘤能力以及HBV复制。从机制上看,TRMT6介导的m1A修饰增强了周期蛋白依赖性激酶9(CDK9)mRNA的稳定性和翻译效率。升高的CDK9通过上调其下游致癌效应因子促进HCC进展,并通过使TARDBP蛋白Ser254位点磷酸化,增强pgRNA转录并抑制pgRNA剪接,从而刺激HBV复制。CDK9抑制剂FIT-039可消除这些效应,且无明显毒性。因此,TRMT6介导的m1A修饰双重驱动HCC恶性进展和HBV复制,是一个有前景的治疗靶点,而抑制CDK9可能构成HBV相关HCC的有效治疗策略。本文于2026年4月发表于《Advancedscience》, IF 14.1.
图形摘要
主要实验结果
1. 单细胞核转录组图谱显示HCC组织中m1A修饰水平及TRMT6表达升高
我们对4例HBV阳性HCC组织和2例配对癌旁组织进行了snRNA-seq,并整合了GSE242889数据集中另外5例癌旁组织进行综合分析(图1A)。共计纳入76634个细胞进行后续研究,去除了重复细胞和质量较差的细胞。我们采用统一流形逼近与投影(UMAP)对人群肝细胞群体进行可视化。基于各细胞类型特异性标志物的表达矩阵,我们鉴定出9种主要细胞类型,包括成纤维细胞(FC)、肝细胞(HEP)、内皮细胞(EN)、双细胞(Doubled)、增殖细胞(PC)、髓系细胞(MC)、上皮细胞(EPI)、B细胞(BC)和T/NK细胞(T_NK)。所有细胞类型在肿瘤和对照组织中的比例见图S1F。
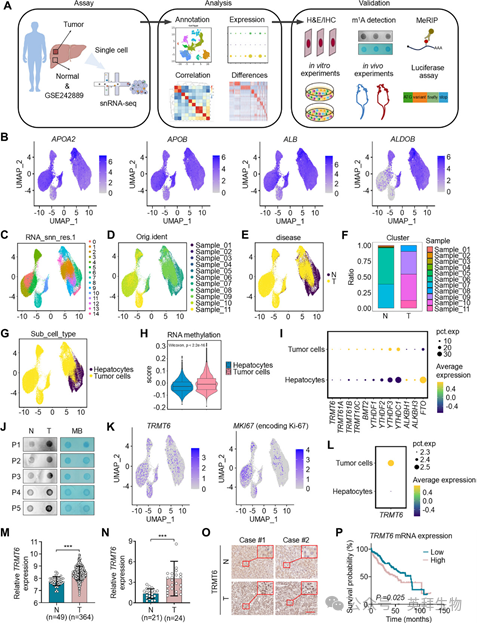
图1 通过snRNA-seq分析人HCC及非癌组织鉴定细胞类型
接下来,我们提取了40674个肝细胞进行进一步分析。经过数据降维,我们使用UMAP对人群肝细胞群体进行可视化,并将细胞进一步分为15个亚群(图1B、C)。采用Harmony方法进行数据整合,以去除批次效应并确保一致性和可比性(图1D、E)。根据特异性标志物的表达,细胞被定义为肝细胞和肿瘤细胞(图1F、G)。随后,我们分析了肝细胞和肿瘤细胞中RNA甲基化相关基因集的评分,发现肿瘤细胞中的RNA甲基化水平高于肝细胞(图1H)。为了探讨m1A修饰在HCC中的作用,我们分析了m1A相关分子的基因表达,发现m1A写入器和读取器在肿瘤细胞中的表达高于肝细胞,而擦除器的表达则相反,在肿瘤细胞中低于肝细胞(图1I)。这些结果表明m1A修饰与HCC的发生或进展相关。
为了验证上述结果,我们通过斑点印迹法检测了5对HCC和对照组织中m1A的水平,发现HCC组织中m1A水平显著升高(图1J)。作为主要的m1A写入器,TRMT6在肿瘤细胞中高表达,这与增殖标志物MKI67的表达模式一致(图1K、L)。接下来,我们利用癌症基因组图谱(TCGA)数据库分析了TRMT6的mRNA水平。非配对(图1M)和配对分析均一致显示,与对照组相比,HCC中TRMT6的表达显著升高。我们进一步评估了新鲜冷冻HCC组织、石蜡包埋HCC切片及其对照组织中TRMT6的mRNA和蛋白水平。qRT-PCR(图1N)和免疫组织化学(IHC)(图1O)的结果进一步支持了上述结论。Kaplan-Meier生存分析也表明,TRMT6高表达与HCC患者的不良生存显著相关(图1P)。综合这些数据表明,TRMT6介导的m1A修饰可能在HCC中发挥致癌作用。
2. TRMT6介导的m1A修饰促进HCC细胞的恶性表型
为探究TRMT6介导的m1A修饰对HCC的作用,我们在MHCC97H和Huh7细胞中敲低并异位表达了TRMT6(图2A、B,左图)。TRMT6敲低细胞中m1A水平降低(图2A,右图),而TRMT6过表达细胞中m1A水平升高(图2B,右图),进一步证实了TRMT6对m1A修饰的调控作用。接下来,我们评估了TRMT6对细胞活力的影响。结果显示,敲低TRMT6显著削弱了HCC细胞的增殖能力(图2C)和集落形成能力(图2D)。此外,流式细胞术分析显示,敲低TRMT6后凋亡细胞显著增加(图2E),并且TRMT6敲低明显诱导了G0/G1期细胞周期阻滞(图2F)。Transwell实验表明,敲低TRMT6显著抑制了HCC细胞的迁移和侵袭能力(图2G)。相反,TRMT6过表达则显著增强了细胞的增殖和集落形成能力。
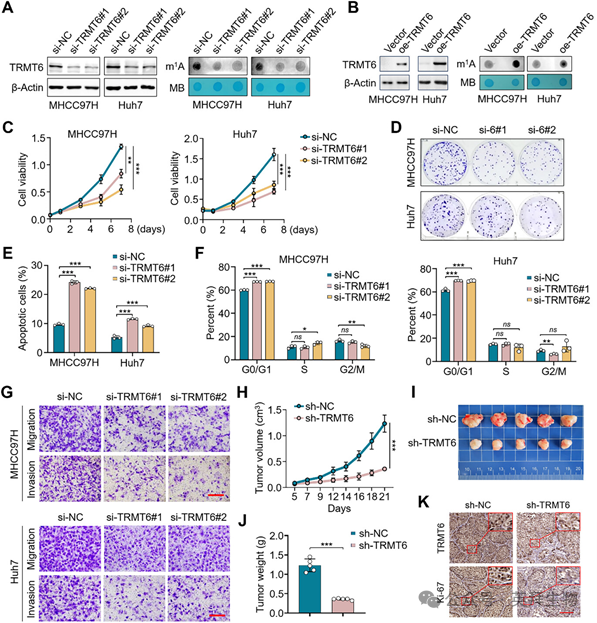
图2 TRMT6介导的m1A修饰增强HCC细胞的恶性行为
为了进一步评估TRMT6介导的m1A修饰对HCC细胞体内致瘤性的影响,我们采用慢病毒系统在MHCC97H细胞中稳定敲低TRMT6,并将TRMT6敲低细胞及其对照细胞皮下接种到裸鼠体内,构建异种移植瘤模型。结果显示,与对照肿瘤相比,来源于TRMT6敲低细胞的异种移植瘤生长更慢(图2H),重量更轻(图2I、J)。此外,移植瘤的Ki-67染色证实了TRMT6敲低细胞的增殖能力下降(图2K)。综上所述,这些数据支持TRMT6介导的m1A修饰在HCC中发挥致癌作用。
3. TRMT6介导的m1A修饰促进HBV复制
HBV感染是HCC的主要危险因素,且HCC的发病率随血清HBV DNA水平升高而增加。考虑到有效的抗病毒治疗可以降低HCC的复发率并改善HBV相关HCC患者的预后,我们试图确定TRMT6介导的m1A修饰在调控HBV复制中的潜在作用。根据HBV感染状态,将细胞分为HBV阳性(HBV+)、HBV阴性(HBV-)和未知(NA)亚组(图3A)。随后我们分析了HBV+和HBV-亚组之间m1A相关调控因子的表达。与HBV-亚组相比,HBV+亚组中大多数m1A写入器和读取器表达上调,而m1A擦除器则呈现相反的表达模式(图3B)。其中,TRMT6在高滴度HBV组织中的表达水平显著高于低滴度HBV组织和癌旁非癌组织(图3C)。我们还分析了新鲜冷冻或石蜡包埋的HBV+ HCC样本及其对照组织中TRMT6的mRNA和蛋白水平。qRT-PCR(图3D)和IHC(图3E)的结果进一步证实TRMT6在HBV+ HCC样本中表达升高。Kaplan-Meier生存分析表明,TRMT6高表达与HBV+ HCC患者的不良预后相关(图3F)。
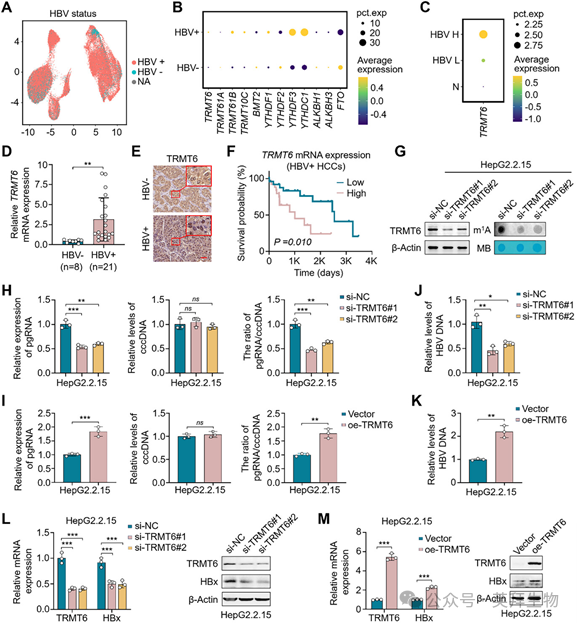
图3 TRMT6介导的m1A修饰对HBV复制的促进作用
接下来,我们在HepG2.2.15细胞(该细胞基因组中整合了两份头尾相连的HBV DNA序列)中敲低了TRMT6(图3G,左图),并如预期发现,与对照细胞相比,TRMT6敲低细胞中的m1A水平显著降低(图3G,右图)。鉴于pgRNA转录对HBV复制至关重要,甚至决定其复制速率,我们检测了pgRNA和HBV cccDNA的水平,以确定TRMT6敲低对HepG2.2.15和HepAD38细胞中pgRNA转录效率(pgRNA/cccDNA)的影响。结果表明,TRMT6敲低显著降低了pgRNA的表达(图3H,左图),但不影响HBV cccDNA水平(图3H,中图),因此降低了pgRNA的转录效率(图3H,右图)。相反,异位表达TRMT6则提高了pgRNA的转录效率(图3I,右图),表现为pgRNA水平升高(图3I,左图)而HBV cccDNA水平不变(图3I,中图)。此外,TRMT6敲低降低了释放到细胞上清中的病毒颗粒内HBV DNA的含量(图3J),而TRMT6过表达则增加了HBV DNA水平(图3K)。如预期,TRMT6敲低细胞中HBx的mRNA和蛋白水平均显著降低(图3L),而在TRMT6过表达细胞中则升高(图3M)。这些数据表明,TRMT6介导的m1A修饰促进了HBV复制,从而增加了HCC的致癌风险。
4. TRMT6在HCC中上调CDK9的表达
鉴于TRMT6参与mRNA的m1A修饰,我们接下来探究TRMT6是否通过调控主要癌基因mRNA的m1A修饰来促进HCC细胞的恶性表型。为了鉴定受m1A修饰调控的mRNA转录本,我们分析了三个m1A-RIP-seq数据集(这些数据集来自HepG2和HeLa细胞中m1A的全基因组图谱、TRMT6/TRMT61A过表达后积累的m1A位点以及H2O2诱导的m1A峰),以及两个来自YTHDF2和YTHDC1结合m1A位点的PAR-CLIP数据集。结果发现,在所有数据集中均一致鉴定出的基因只有周期蛋白依赖性激酶9(CDK9)和线粒体核糖体蛋白L4(MRPL4)(图4A)。MRPL4是线粒体核糖体的关键组分,但在人类恶性肿瘤(尤其是HCC)中的特征尚不明确。然而,我们观察到敲低TRMT6后,MRPL4的mRNA和蛋白水平均未发生变化。CDK9作为一种典型的转录CDK,已被广泛证实在包括HCC在内的多种恶性肿瘤中具有致癌活性。也有报道称其抑制剂可抑制HBV复制。IHC染色结果显示,与对照组织相比,HCC组织中CDK9的表达升高(图4B),这支持了上述观点。我们使用TCGA数据库进一步验证了这些结果。生存分析显示,CDK9高表达与HCC患者50个月无病生存期缩短显著相关。值得注意的是,这种不良预后相关性在HBV阳性HCC患者中更为显著。
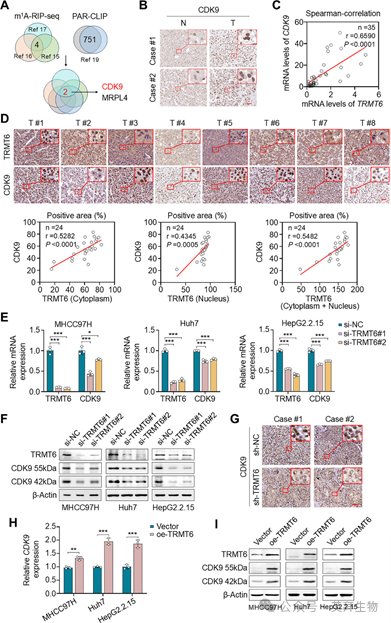
图4 TRMT6上调CDK9表达
我们随后发现,在HCC中CDK9与TRMT6的mRNA表达水平呈显著正相关(图4C)。与之对应的是,在24例石蜡包埋的HCC样本中,CDK9的蛋白水平与定位于细胞质、细胞核及全细胞中的TRMT6均呈正相关(图4D),进一步验证了上述结论。为了确定TRMT6对CDK9的调控作用,我们在MHCC97H、Huh7和HepG2.2.15细胞中敲低TRMT6,发现敲低TRMT6导致CDK9的mRNA(图4E)和蛋白(图4F)水平显著降低,CDK9存在分子量分别为55 kDa和42 kDa的两种亚型。这一观察结果在异种移植瘤组织的CDK9 IHC染色中得到了证实(图4G)。相反,在这些细胞中异位过表达TRMT6则上调了CDK9的mRNA(图4H)和蛋白(图4I)水平。为了排除m6A修饰可能参与调控CDK9表达,我们用METTL3抑制剂STM2457处理MHCC97H、Huh7和HepG2.2.15细胞,结果未能像敲低TRMT6后那样观察到CDK9表达的一致下调。总之,这些数据表明TRMT6可能以m1A依赖的方式调控CDK9表达。
5.TRMT6介导的CDK9 mRNA的m1A修饰增强其稳定性和翻译效率
接下来,我们使用RMBase v2.0数据库(https://rna.sysu.edu.cn/rmbase/m1Amod.php)预测了CDK9 mRNA中的m1A基序,并在其编码DNA序列(CDS)中发现了一个m1A基序“GTTCGA”(图5A)。甲基化RNA免疫沉淀(MeRIP)结合qRCR(MeRIP-qPCR)可用于检测m1A修饰水平。为此,我们首先设计了两对引物,分别检测CDK9 mRNA中m1A上游序列“U”和包含m1A的序列“M”(图5B)。MeRIP-qPCR结果显示,在MHCC97H、Huh7和HepG2.2.15细胞中异位表达TRMT6显著增加了m1A水平(图5C)。
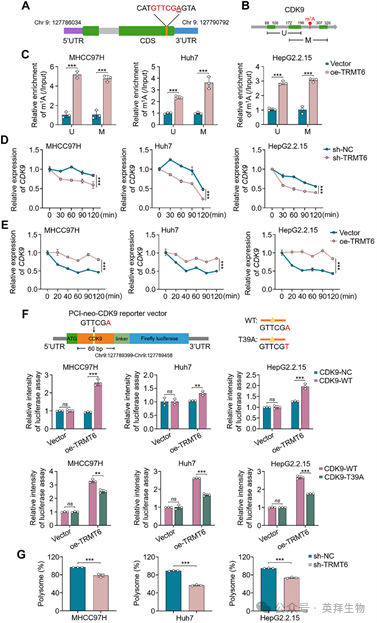
图5 TRMT6介导的m1A修饰增强CDK9 mRNA的稳定性和翻译效率
RNA甲基化修饰(如m6A和m1A)通常影响mRNA的稳定性和翻译效率。因此,我们用10 µM放线菌素D(ACTD)处理TRMT6敲低或过表达的MHCC97H、Huh7和HepG2.2.15细胞及其对照细胞,以抑制mRNA转录,然后评估它们对CDK9 mRNA稳定性的影响。结果表明,与对照相比,敲低TRMT6显著加速了CDK9 mRNA的降解(图5D),而TRMT6过表达则增强了CDK9 mRNA的稳定性(图5E)。我们还基于pCI-neo载体构建了包含野生型m1A基序(GTTCGA)或点突变型m1A基序(GTTCGT)的荧光素酶报告质粒(图5F,上图),分别命名为CDK9-WT和CDK9-T39A。结果显示,TRMT6过表达显著增加了CDK9-WT报告质粒的萤火虫荧光素酶强度(图5F,中图),而CDK9-T39A则显著减弱了这一效应(图5F,下图),表明m1A修饰促进了CDK9 mRNA的翻译。与之相符的是,我们通过多聚体谱分析证明,在MHCC97H、Huh7和HepG2.2.15细胞中敲低TRMT6显著降低了CDK9 mRNA在多聚体层中的分布(图5G)。接下来,我们通过siRNA介导的敲低筛选了多个已知的m1A读取器。结果显示,在所检测的三个细胞系中,这些已知读取器均未显著调控CDK9的mRNA或蛋白水平。这些结果使我们合理推断,介导CDK9调控的m1A读取器可能不在目前已鉴定的读取器之中,而可能是一种在mRNA稳定性和翻译调控中具有双重作用的新型读取器。综上所述,这些数据表明,TRMT6介导的CDK9 mRNA的m1A修饰增强了其稳定性和翻译效率。
6.抑制CDK9可消除TRMT6对HCC细胞恶性行为及HBV复制的促进作用
为了进一步明确TRMT6介导的mRNA m1A修饰通过上调CDK9在HCC中发挥的致癌功能,我们首先用选择性CDK9抑制剂FIT-039处理MHCC97H和Huh7细胞,结果显示FIT-039在MHCC97H细胞中的IC50值为22.13 µM,在Huh7细胞中为13.93 µM。同时,我们观察到FIT-039处理显著逆转了TRMT6过表达所诱导的促增殖和促集落形成效应(图6A、B)。一致地,在过表达TRMT6的MHCC97H和Huh7细胞中敲低CDK9同样减弱了TRMT6对细胞增殖和集落形成的增强作用。此外,FIT-039处理和CDK9沉默均能有效消除TRMT6过表达对细胞凋亡的抑制作用(图6C)。
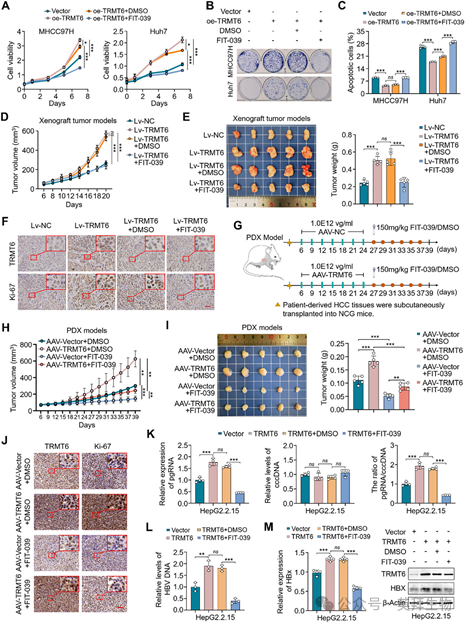
图6 TRMT6介导的CDK9上调促进HCC细胞的恶性表型及HBV复制
为了进一步研究TRMT6介导的CDK9上调对HCC细胞体内致瘤性的影响,我们利用慢病毒系统在MHCC97H细胞中稳定过表达TRMT6,并将TRMT6过表达细胞及其对照细胞皮下接种到裸鼠体内。随后,对携带TRMT6过表达细胞来源异种移植瘤的小鼠给予150 mg/kg FIT-039或等剂量的DMSO。结果显示,与对照肿瘤相比,TRMT6过表达细胞来源的异种移植瘤生长更快、重量更大,而FIT-039可有效逆转这一现象(图6D、E)。上述肿瘤中核增殖标志物Ki-67的IHC染色支持了这一结果(图6F)。此外,在整个治疗期间,各实验组之间的小鼠体重未见显著差异。同样,这些小鼠的血清标志物以及肝、肾功能的组织形态学也未发生变化。总之,这些结果表明FIT-039给药不会引起明显毒性。
为了提高我们研究结果的临床相关性和转化潜力,我们通过将人HCC组织皮下植入NCG小鼠体内构建了HCC患者来源异种移植模型(PDX),并通过瘤内注射腺相关病毒(AAV)过表达TRMT6。随后,通过腹腔注射给予荷瘤小鼠150 mg/kg FIT-039或等剂量的DMSO(图6G)。结果显示,在PDX模型中,药物抑制CDK9显著抑制了肿瘤生长,表现为肿瘤体积和重量减小(图6H、I)。Ki-67的IHC染色进一步支持了上述结果(图6J)。在整个治疗期间,所有实验组的小鼠体重保持相当。此外,血清生化指标以及肝脏和肾脏切片的H&E染色表明,FIT-039治疗未引起明显毒性。
接下来,我们用10 µM FIT-039处理过表达TRMT6的HepG2.2.15细胞,以评估其对HBV复制的影响。同时,在过表达TRMT6的HepAD38细胞中敲低CDK9,以排除FIT-039的潜在脱靶效应。结果显示,FIT-039和CDK9沉默均有效逆转了TRMT6介导的pgRNA转录效率升高(图6K,右图),表现为pgRNA水平恢复(图6K,左图)而cccDNA水平不变(图6K,中图)。此外,FIT-039或CDK9敲低同样能够消除TRMT6过表达对HBV DNA(图6L)和HBx(图6M)水平的刺激作用。综上所述,我们的研究结果支持TRMT6介导的m1A修饰通过上调CDK9促进HCC进展和HBV复制,凸显了它们作为HBV相关HCC治疗靶点的潜力。
7. CDK9通过上调其下游致癌效应因子增强HCC细胞的恶性表型
作为一种丝氨酸/苏氨酸(Ser/Thr)激酶,CDK9通过上调关键的下游效应因子发挥其致癌活性。因此,我们在MHCC97H、Huh7和HepG2.2.15细胞中研究了沉默或过表达TRMT6对CDK9关键致癌下游靶点水平的影响,这些靶点包括磷酸化的STAT3(p-STAT3)、MCL1和BCL-2,它们被广泛认为是HCC发病机制中的关键介质。结果如预期所示,敲低TRMT6显著降低了这些致癌蛋白的水平(图7A),这一结果在TRMT6敲低异种移植瘤和对照肿瘤的IHC染色中得到了进一步验证(图7B)。相反,过表达TRMT6提高了p-STAT3、MCL1和BCL-2的水平(图7C),而这一效应可被FIT-039和CDK9敲低所逆转(图7D)。这些观察结果在来自图6E、I的异种移植瘤中这些分子的IHC染色中得到了进一步证实(图7E)。这些数据共同表明,TRMT6介导的CDK9上调通过激活其核心促肿瘤下游效应因子来驱动HCC进展。
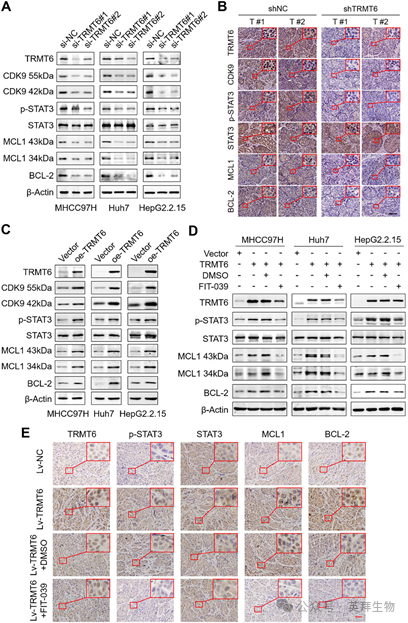
图7 TRMT6通过CDK9介导的致癌效应因子上调在HCC中发挥致癌作用
8. CDK9通过磷酸化TARDBP的Ser254位点促进HBV复制
尽管抑制CDK9可以阻碍HBV复制,但其潜在的分子机制仍不明确。鉴于CDK9主要通过与底物蛋白相互作用并使其磷酸化来发挥激酶功能,我们在HepG2.2.15和HepG2细胞中进行了免疫共沉淀(Co-IP)联合液相色谱-串联质谱(LC-MS/MS)分析,以鉴定潜在的CDK9底物。对HepG2.2.15细胞中候选蛋白的基因本体论(GO)分析揭示了15个与病毒过程和生命周期相关的条目。通过比较HepG2.2.15和HepG2细胞之间的差异独特肽段,我们进一步分析了参与病毒过程的表达上调和下调的前10个分子。其中,TAR DNA结合蛋白(TARDBP,也称TDP-43)是一种核DNA/RNA结合蛋白,参与mRNA转录、剪接、稳定性和翻译调控,此前已发现与HBV复制相关。LC-MS/MS鉴定出的其肽段序列见图S28。因此,选择TARDBP用于后续实验的深入机制研究。
我们首先通过癌症基因组图谱(TCGA)数据库分析了TARDBP的mRNA水平,结果显示与对照组织相比,HCC组织中TARDBP的表达显著升高。随后的生存分析表明,TARDBP表达升高与HCC患者的不良临床结局密切相关,且这种关联在HBV阳性HCC病例中更为显著(图8A)。此外,我们通过反向Co-IP实验验证了HepG2.2.15细胞中CDK9与TARDBP之间的相互作用(图8B)。接下来,我们利用CRISPR-Cas9系统在HepG2.2.15和HepG2 NTCP细胞中敲除了TARDBP。HepG2 NTCP细胞是通过在HepG2细胞中异位表达NTCP建立的,TARDBP成功缺失的验证见图S30C。敲除TARDBP导致pgRNA水平显著降低(图8C)。此外,过表达CDK9(图8D)或TRMT6(图8E)在对照细胞中显著提高了pgRNA水平,但在TARDBP缺陷细胞中则无此效应,这支持了TRMT6–CDK9轴通过TARDBP促进HBV复制的模型。
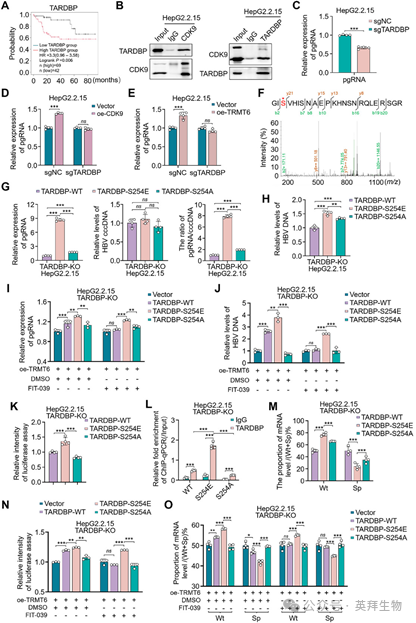
图8 CDK9通过磷酸化TARDBP的Ser254位点促进HBV复制
接下来,我们研究了CDK9如何通过TARDBP调控HBV复制。鉴于其激酶活性,我们假设CDK9直接磷酸化作为底物的TARDBP。为了验证这一点,我们用CDK9抑制剂FIT-039或DMSO处理HepG2.2.15细胞,然后通过Co-IP联合LC-MS/MS鉴定受CDK9调控的TARDBP磷酸化位点。结果显示,FIT-039显著降低了TARDBP在多个丝氨酸残基(包括Ser254)上的磷酸化水平(图8F)。体外激酶实验进一步证实,CDK9直接磷酸化TARDBP的Ser254位点,而S254A突变体则消除了这一修饰。TARDBP的RRM2结构域(第192–265位氨基酸)可与HBV核心启动子(CP)结合,这对pgRNA转录至关重要,且Ser254位点对TARDBP的RNA结合亲和力具有关键作用。综上所述,这些发现提示TARDBP Ser254磷酸化可能促进HBV复制。
为了验证上述假设,我们构建了野生型TARDBP(TARDBP-WT)及其磷酸化模拟突变体(S254E)和磷酸化缺陷突变体(S254A),每个突变体在sgTARDBP识别序列内均携带同义密码子突变。然后将这些构建体转染到TARDBP敲除的HepG2.2.15和HepG2 NTCP细胞中,并评估它们对HBV复制的影响。结果显示,与TARDBP-WT相比,TARDBP S254E显著增加了pgRNA水平,而与TARDBP S254E相比,TARDBP S254A则显著降低了pgRNA丰度(图8G,左图)。两种突变体均未改变cccDNA水平(图8G,中图),它们对pgRNA转录效率的影响与其对pgRNA水平的影响一致(图8G,右图)。同样,与TARDBP-WT相比,TARDBP S254E显著提高了HBV DNA和HBx的水平,而TARDBP S254A则消除了这些效应(图8H)。为了进一步验证这些结果,我们在TARDBP敲除的HepG2.2.15细胞中异位表达TRMT6,并同时加入10 µM FIT-039。结果表明,TARDBP WT提高了pgRNA和HBV DNA的水平,TARDBP S254E进一步增强而TARDBP S254A减弱了这些效应(图8I、J)。值得注意的是,在FIT-039处理后,仅有TARDBP S254E仍保留强烈促进pgRNA和HBV DNA水平的能力(图8I、J)。这些数据表明,CDK9介导的TARDBP Ser254磷酸化促进了HBV复制。
9. TARDBP Ser254磷酸化激活HBV核心启动子并抑制pgRNA剪接
基于上述发现,我们推测TARDBP Ser254磷酸化可能通过增强核心启动子(CP)活性和抑制pgRNA剪接来促进HBV复制。为了验证这一点,在TARDBP敲除的HepG2.2.15和HepG2-NTCP细胞中异位表达TARDBP-WT、-S254E和-S254A,然后使用含有HBV CP区域(nt1613–nt1849)的双荧光素酶报告基因检测CP活性。结果显示,与TARDBP-WT相比,TARDBP-S254E显著增强了萤火虫荧光素酶强度,而与TARDBP-S254E相比,TARDBP-S254A则显著降低了该强度(图8K)。ChIP-qPCR实验进一步证实,TARDBP-S254E与HBV CP的结合增强,而TARDBP-S254A则显著减弱了这种结合(图8L)。鉴于已知TARDBP可调控pgRNA剪接,我们通过在TARDBP敲除的HepG2.2.15和HepG2-NTCP细胞中异位表达TARDBP-WT、-S254E和-S254A,检测了TARDBP S254磷酸化对野生型(Wt)和缺失1.3 kb内含子的2.2 kb剪接变体(Sp)pgRNA水平及比例的影响。结果显示,与TARDBP-WT相比,TARDBP-S254E显著提高了pgRNA中Wt的比例并降低了Sp的比例,而TARDBP-S254A则显著减弱了这一效应(图8M)。接下来,我们在TARDBP敲除细胞中异位表达TRMT6,并用FIT-039处理这些细胞,评估它们对HBV CP活性和pgRNA剪接的影响。与对照组相比,TARDBP-WT显著增加了萤火虫荧光素酶强度(图8N)和Wt pgRNA的比例(图8O),TARDBP-S254E进一步增强而TARDBP-S254A则显著降低了这一效应。综上所述,这些结果证明TRMT6-CDK9轴驱动TARDBP Ser254磷酸化,从而通过刺激pgRNA转录和抑制pgRNA剪接来促进HBV复制。
通过总结上述发现,我们阐明了TRMT6介导的m1A修饰通过上调CDK9促进HCC进展和HBV复制的机制。具体而言,TRMT6表达增加通过m1A修饰增强CDK9 mRNA的稳定性和翻译效率,从而上调其表达。CDK9一方面通过上调其下游致癌效应因子(如p-STAT3、MCL1和BCL-2)的水平促进HCC细胞的恶性表型;另一方面,CDK9通过磷酸化TARDBP的Ser254位点,激活HBV CP以促进pgRNA转录,并抑制pgRNA剪接,从而促进HBV复制并增加HCC的风险。
参考文献:
Zhang R, Zong D, Liu R, Wang Y, Gu Q, Yao Y, Zheng W, Yuan M, Wang S, Cui R, Li D, Dang S, Hou P. TRMT6-Mediated m1A Modification of CDK9 mRNA Is a Dual-Pronged Pathogenic Driver for HBV-Related Hepatocellular Carcinoma. Adv Sci (Weinh). 2026 Apr 20:e14172. doi: 10.1002/advs.202514172. Epub ahead of print. PMID: 42003777.