






看METTL3介导的m6A如何作用于胃癌的上皮间质转化和转移
N6-甲基腺苷(m6A)修饰是真核mRNA中最常见的化学修饰之一,对mRNA的稳定性,剪接和翻译产生重要影响。最近,人们越来越认识到m6A在肿瘤发生中的调节作用。然而,m6A的失调及其在肿瘤上皮-间质转化(EMT)和转移中的功能仍然不清楚。该研究采用qRT-PCR和免疫组织化学方法检测胃癌(GC)中METTL3的表达。通过体外和体内试验研究了METTL3对GC转移的影响。通过转录组测序,m6A测序,m6A甲基化RNA免疫定量逆转录聚合酶链反应(MeRIP qRT-PCR),共聚焦免疫荧光测定,荧光素酶报告基因测定,共免疫沉淀,RNA免疫沉淀和染色质免疫沉淀测定等方法探索了METTL3的作用机理。

结果:
一、METTL3过表达及其在GC中的预后价值
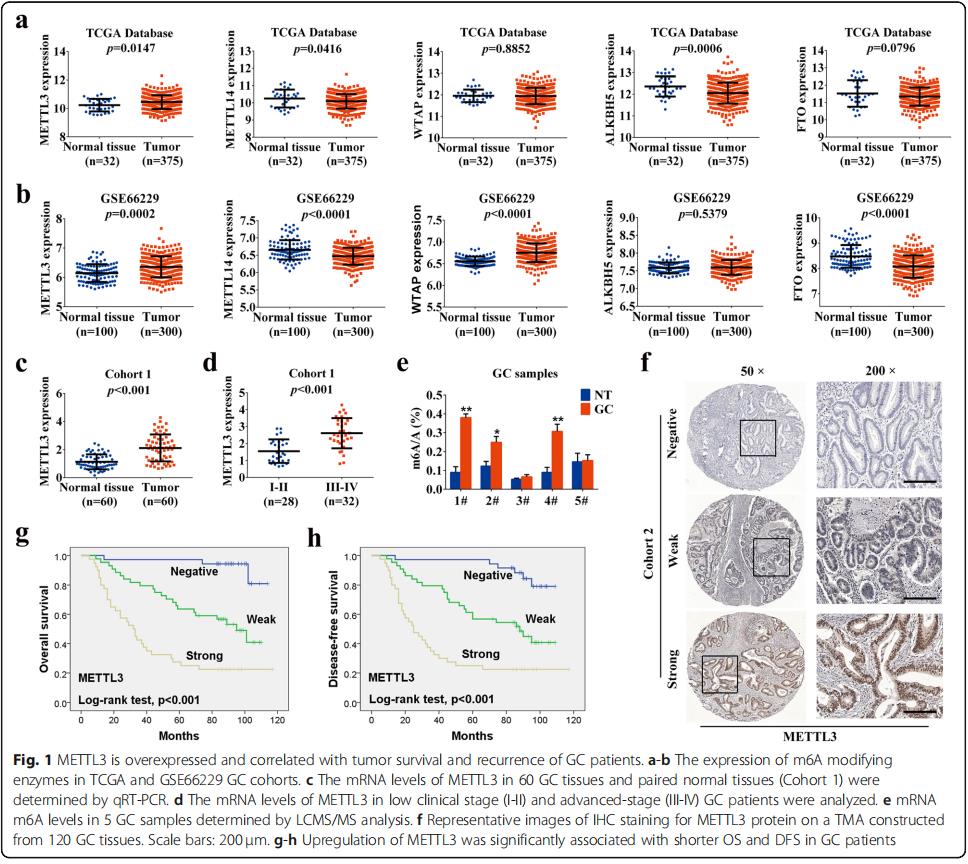
通过临床数据集TCGA(癌症基因组图谱)和GSE66229,发现GC组织中METTL3 mRNA表达显着升高,胃癌组织中METTL14表达降低,且两个数据集中的其他m6A“writers” 和 “erasers”水平不一致(图1a,b)。qRT-PCR验证了由60对GC组织中METTL3的mRNA水平明显过表达(图1c),METTL3在晚期(III-IV)的GC组织中明显更高(图1d),GC组织中观察到m6A mRNA的水平升高(图1e)。免疫组化检查了120例的GC组织微阵列样品中METTL3的蛋白水平。METTL3显着地定位在GC细胞核中(图1f)。Kaplan-Meier曲线显示,具有METTL3高表达的患者表现出较差的总生存期(OS)和无病生存期(DFS)(图1g,h)。总之,METTL3在GC中明显过表达,可能与GC进展有关。
二、EMT需要METTL3
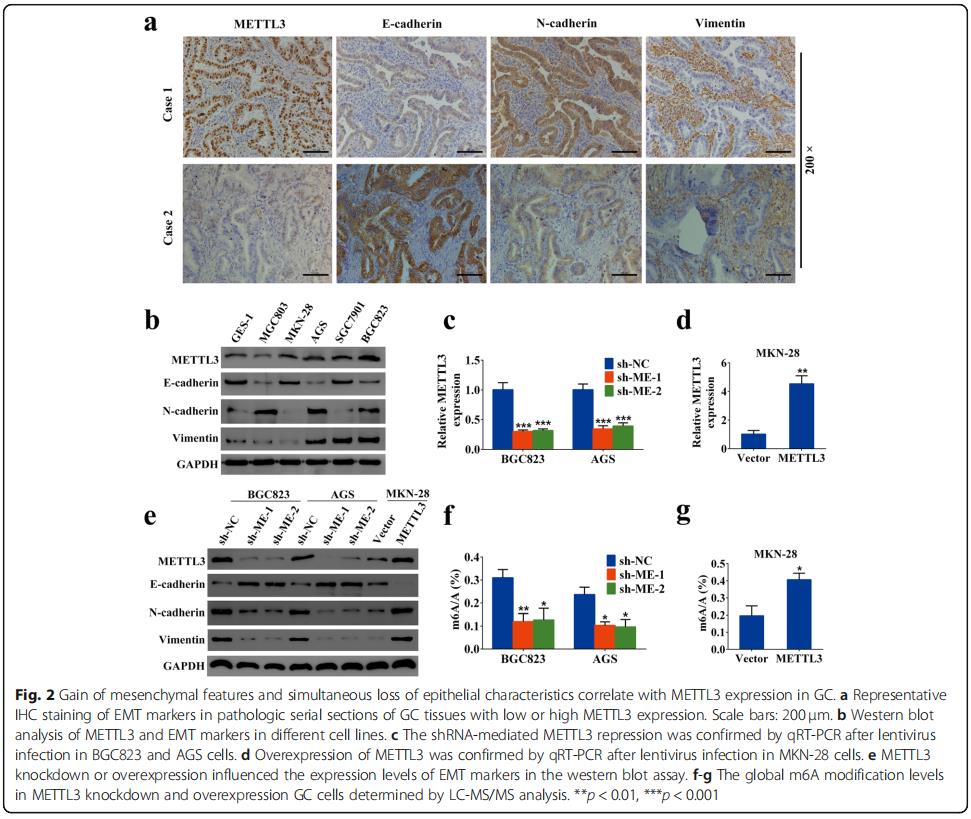
免疫组化连续切片染色显示GC组织中METTL3的表达水平似乎与E-钙粘蛋白负相关,而与N-钙粘蛋白和波形蛋白正相关(图2a)。MKN-28细胞系显示E-钙粘蛋白的高表达,而N-钙粘蛋白和波形蛋白的水平低,这是其上皮表型的代表(图2b)。为验证EMT程序是否需要METTL3,进行了功能丧失和功能获得研究(图2c,d)。敲低METTL3会导致N-钙粘蛋白和波形蛋白的显着下调,同时在BGC823和AGS细胞中蛋白质水平上E-钙粘蛋白的显着上调。METTL3的过表达在MKN-28细胞中引起相反的结果(图2e),METTL3的敲低或过表达分别也降低或增加了GC细胞中总体m6A修饰水平(图2f,g)。总之,METTL3促进了GC细胞中的EMT程序。
三、METTL 3促进胃癌细胞在体内外的侵袭和转移
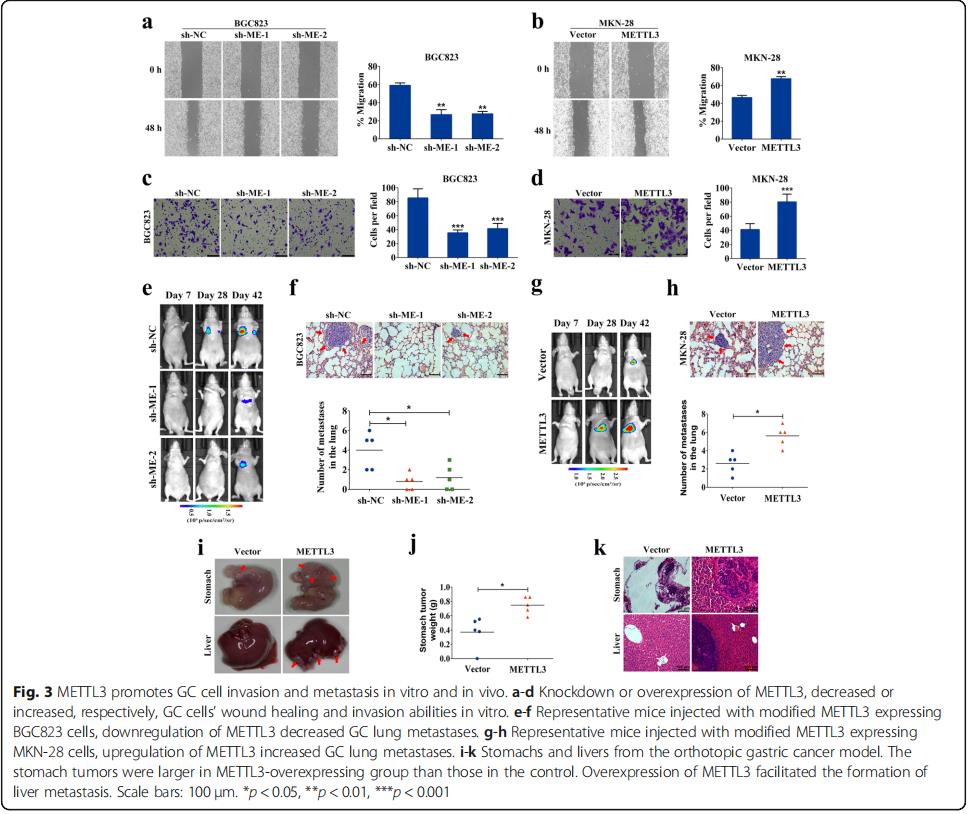
通过伤口愈合实验显示METTL 3表达的减弱明显抑制了BGC 823细胞的迁移能力(图3a),METTL 3的强制表达明显增加了MKN-28细胞的迁移速度(图3b)。Transwell侵袭实验证实,敲除METTL 3基因后,GC细胞的侵袭能力受到明显抑制(图 3C),而METTL 3的异位表达则显着地增强了它的表达(图3d)。研究发现BGC 823-shRNA细胞转移到裸鼠肺部的效果不如对照组(图3e,f)。METTL 3的过表达明显增加了MKN-28细胞的肺转移负担(图3g,h)。小鼠原位模型实验,在裸鼠胃浆膜下注射稳定的MKN-28细胞,GC细胞植入后5周观察肿瘤浸润肌层和粘膜发现METTL 3高表达组的肿瘤比对照组大。此外,METTL 3过表达组可观察到肝转移灶,但不在对照中(图3i-k)。表明METTL3在促进GC细胞侵袭和转移中起着至关重要的作用。
四、转录组-测序和m6A-测序鉴定ZMYM 1是METTL 3的直接靶点
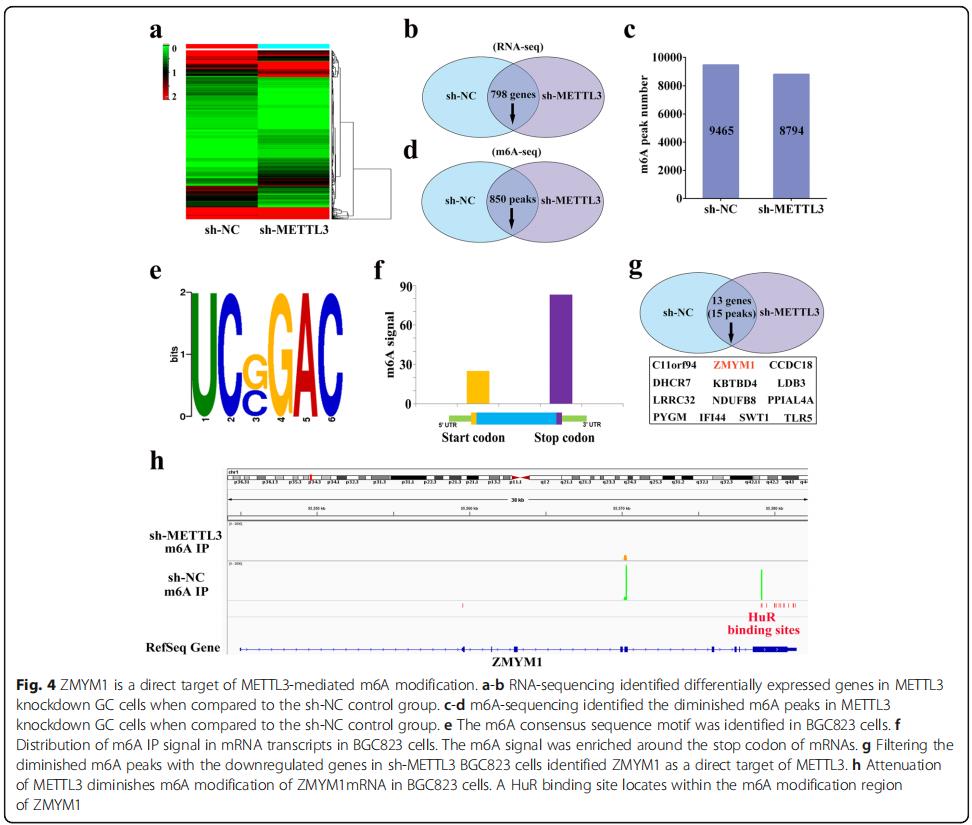
通过转录组测序比较在BGC 823细胞中敲除METTL 3后的基因表达谱,结果发现798个基因被显著下调,662个基因被显著上调(图4a,b)。通过m6A测序鉴定了9465和8794 m6A峰对照和METTL3-缺陷细胞(图4c)。在BGC823细胞中,METTL3稳定敲低,发现850个峰减少(图4d)。在BGC 823细胞中映射m6A甲基体时,在免疫纯化的RNA中,m6A共识序列GGAC(R RACH)基序在m6A位点中高度富集(图4e)。与以前的研究一致,我们证明了m6A信号。 L在mRNAs的终止密码子附近富集(图4f)。用798个下调的基因过滤掉850个减少的m6A峰,从而鉴定出由13个基因带来的15个峰(图4g)。在这13个潜在的调节因子中,我们的重点是ZMYM1,因为在sh-NC BGC823细胞中在其mRNA的终止密码子附近检测到两个m6A峰,并且在METTL3敲低后均被减弱(图4h)。ZMYM 1的生物学功能尚不清楚,因此选择了ZMYM1作为METTL 3介导的m6A修饰的候选靶点。
五、METTL 3在GC中维持ZMYM 11的表达
与基因表达数据一致,分别在mRNA和蛋白水平上ZMYM1的表达被下调或上调的METTL3的敲低或过表达(图5a-d)。免疫荧光染色显示ZMYM 1的核定位及METTL3的缺失诱导BGC 823细胞ZMYM 1的表达损失,而过表达METTL 3则增加MKN-28细胞的ZMYM 1水平(图5e)。qRT-PCR验证了队列1中ZMYM1的表达。 在这个样本人群中,ZMYM1表达经常在GC组织中升高,并与METTL3表达正相关(图5f,g)。 通过共聚焦成像也证实了METTL3和ZMYM1在GC组织中的核共定位(图5h)。数据表明,METTL 3正调控ZMYM 1在GC中的表达。
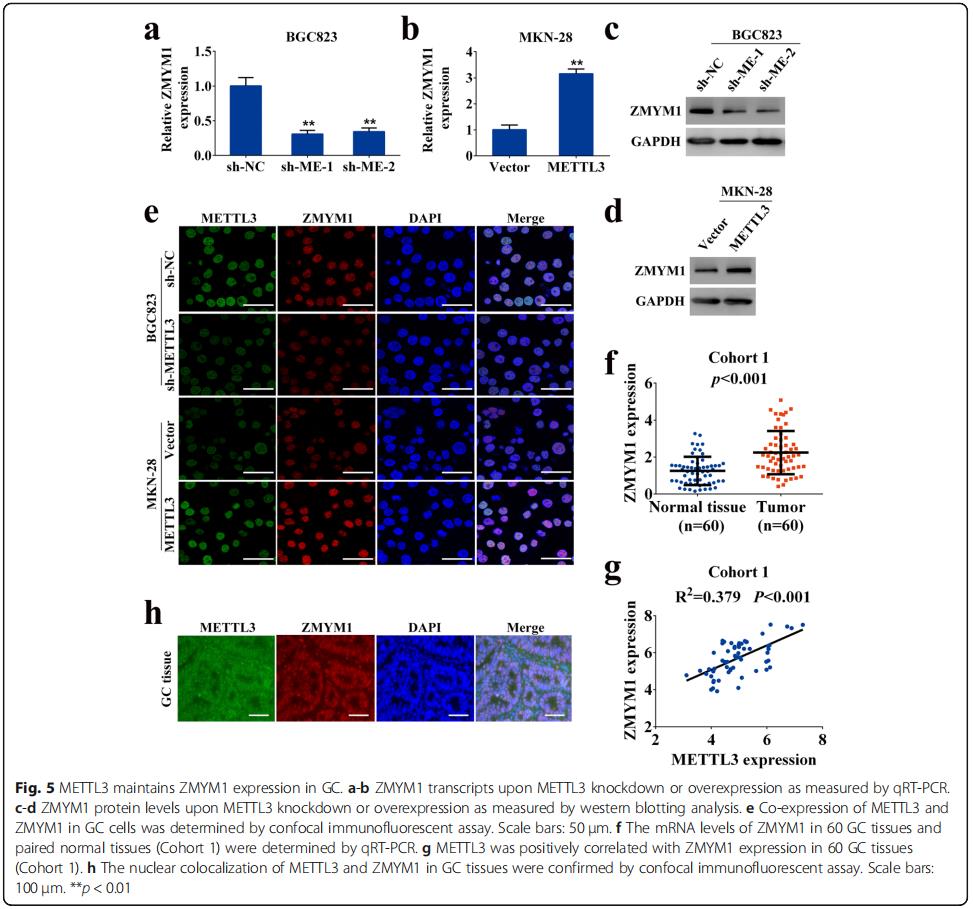
六、METTL 3通过m6A-Hur依赖途径增强ZMYM 1 mRNA表达
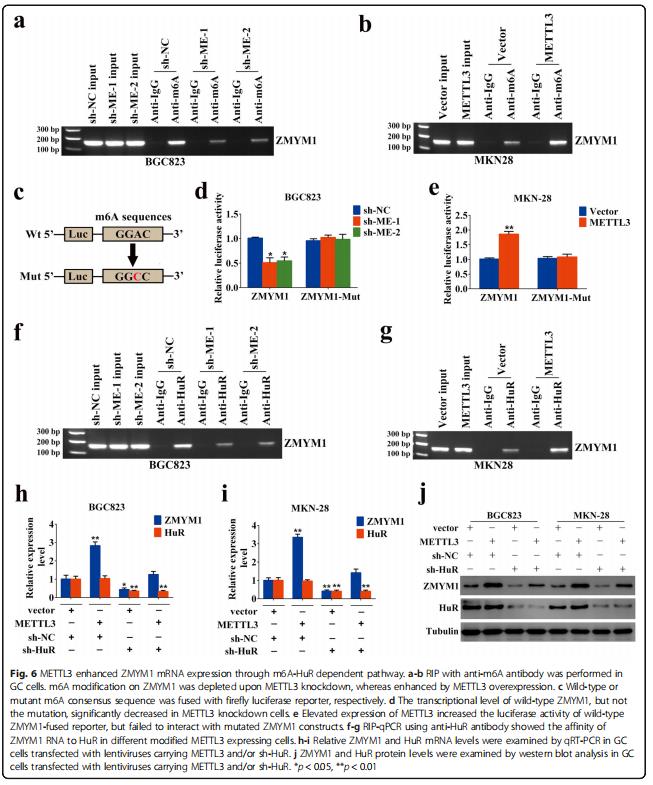
MERIP、qRT-PCR对m6A测序数据集进行了验证,结果抗m6A抗体显著富集胃癌细胞中ZMYM1 mRNA水平的变化。METTL 3的敲除或过表达显著降低或增加了ZMYM1 mRNA的m6A水平(图6a,b)。突变体ZMYM 1的m6A修饰因m6A一致序列(RACH)中的胞嘧啶取代腺苷碱而被取消(图6c)。荧光素酶报告试验表明,野生型ZMYM 1的转录水平不高,突变体在没有METTL 3的情况下显著降低(图6d)。METTL 3过表达增强了野生型ZMYM 1融合报告基因的表达,显示ZMYM 1水平的调控受METTL 3相关m6A修饰的控制(图6e)。HuR结合位点位于ZMYM1的m6A修饰区域,这是由我们的m6A测序所揭示的(图4f)。使用抗HuR抗体的RIP-qPCR显示,在METTL3沉默的BGC823细胞中,HuR对ZMYM1 mRNA的亲和力大大降低,而在MKN-28细胞中,METTL3的过表达则显示出相反的结果(图6f,g)。经修饰的METTL 3表达后,所有GC细胞的Hur表达情况均无明显变化(图6~j)。揭示了METTL 3介导的m6A修饰通过m6A-Hur依赖性途径增强ZMYM 1的表达。
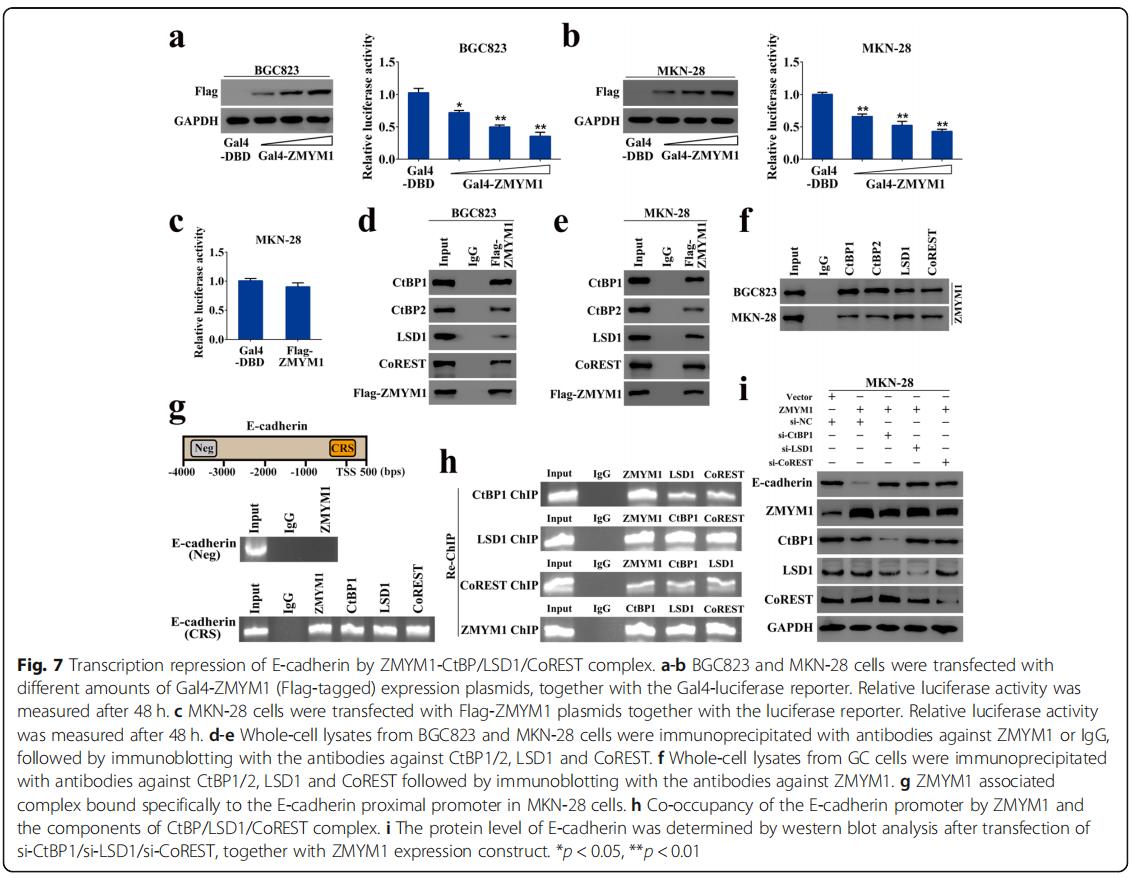
七、ZMYM 1与核内的CtBP/LSD 1/COREST复合物有物理联系
锌指家族蛋白参与了转录调控。我们将全长ZMYM 1融合到gal 4的DNA结合区,并测试了其在GC细胞中的转录活性。结果表明,标记ZMYM1在BGC823和MKN-28细胞中均以剂量依赖性方式显著抑制报告基因的活性(图7a,b)。ZMYM1的过表达根本不影响Gal4驱动的报道分子的活性(图7c),这表明ZMYM1与DNA物理结合,发挥其转录抑制活性。IP用抗ZMYM1的抗体进行IP免疫,然后用抗CtBP / LSD1 / CoREST复合物成分的抗体进行免疫印迹,证明ZMYM1与所有测试的蛋白质都有相互作用(图7d,e)。相反,用抗CtBP1 / 2,LSD1或CoR EST抗体进行IP免疫接种,然后用抗ZMYM1抗体进行免疫印迹也证实ZMYM1被该复合物的所有组分有效地免疫共沉淀(图7f)。结果表明ZMYM1在物理上与CtBP / LSD1 / CoREST复合体相关。
八、ZMYM 1相关复合物对E-钙粘素的转录抑制作用
实验中,对CRS上所有复杂组分的富集进行了验证(图7g)。此外,ZMYM1、CTBP1、LSD1和COREST的共同占有 在E-cadherin启动子上,通过重新芯片分析(图7H)确定了E-cadherin启动子。为了进一步证实ZMYM 1相关复合物对E-cadherin的转录抑制作用,我们进行了WB分析。 裂解并发现ZMYM1的增益-功能伴随E-cadherin的降低表达。显著地,在CTB中,ZMYM1过表达引起的E-cadherin的衰减被取消。 P1,LSD 1,或支柱被击倒(图7i)。数据表明,ZMYM 1通过与CtBP/LSD 1/Corest复合物结合,靶向和抑制E-cadherin的转录。
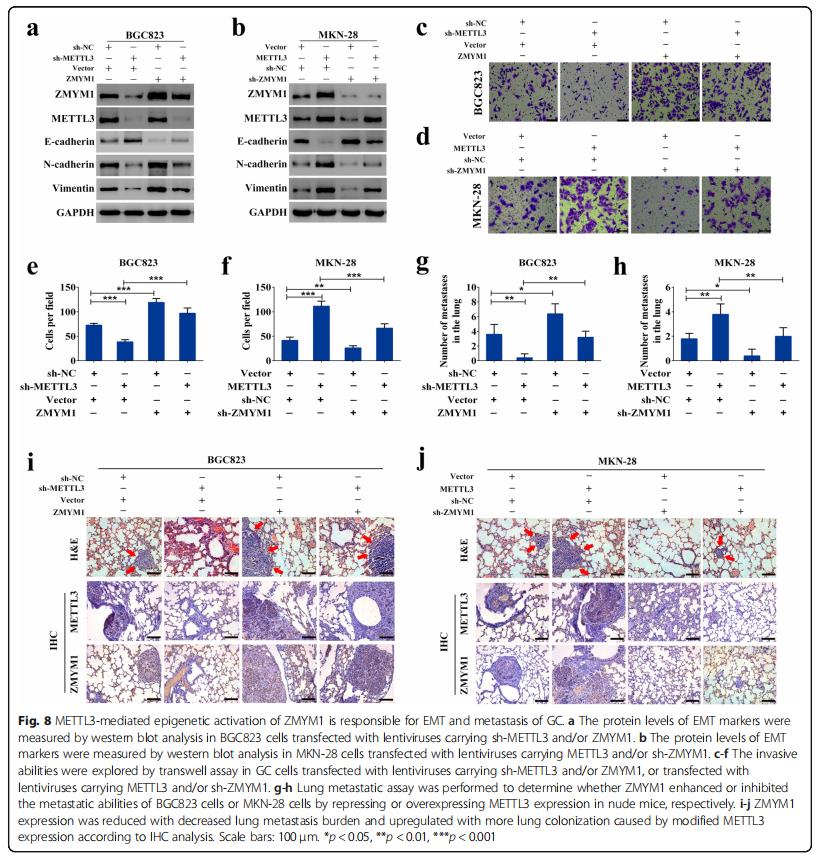
九、METTL 3介导的ZMYM 1表观遗传激活与胃癌的EMT和转移有关
在MYTTL3敲低细胞中,ZMYM1表达的升高概括了N钙粘蛋白和波形蛋白的水平,并降低了E钙粘蛋白的表达(图8a)。相反,抑制ZMYM1至少部分抵消了METTL3过表达引起的EMT进程加速(图8b)。 Transwell入侵试验表明,敲低METTL3降低了GC细胞的侵袭能力,而ZMYM1的异位表达挽救了这种能力(图8c,e)。相反,过表达METTL3可增强细胞侵袭能力,与sh-ZMYM1质粒共转染可部分减弱细胞侵袭能力(图8d,f)。对于肺转移模型,观察到相似的结果。引入ZMYM1可以至少部分抵消METTL3敲低对转移的抑制作用,而与METTL3过表达相关的转移潜能的增加则可以通过ZMYM1的耗尽而部分减弱(图8g,h)。此外,根据免疫组织化学结果,ZMYM1的蛋白水平随着肺转移负担的减少而降低,并因修饰的METTL3表达引起的更多的肺定植而被上调(图8i,j)。表明ZMYM1的过表达与METTL3介导的EMT和转移有关。
结论:
m6A修饰在GC中的关键作用,并发现METTL3 / ZMYM1 / E-钙粘蛋白信号传导是针对GC的抗转移策略的潜在治疗靶标。