






一种新型LncRNA HITT与HIF-1α形成调节环,以调节血管生成和肿瘤生长
导语:
越来越多的证据表明,长的非编码RNA(lncRNA)在包括癌症在内的人类疾病中发挥着重要作用;但是,只有少数几个经过了实验验证和功能注释。在这里,将确定了一种新的lncRNA,称为HITT(翻译水平的HIF-1α抑制剂)。在多种人类癌症中,HITT通常会降低。HITT是缺氧转换后变化最大的lncRNA之一,这篇文章将拓宽我们对lncRNA的癌症相关功能的理解,突出了lncRNA–HIF-1α轴在癌症的靶向治疗中所具有的潜在影响。
技术路线:
1、人类结肠组织样本的获取以及结肠细胞系的培养
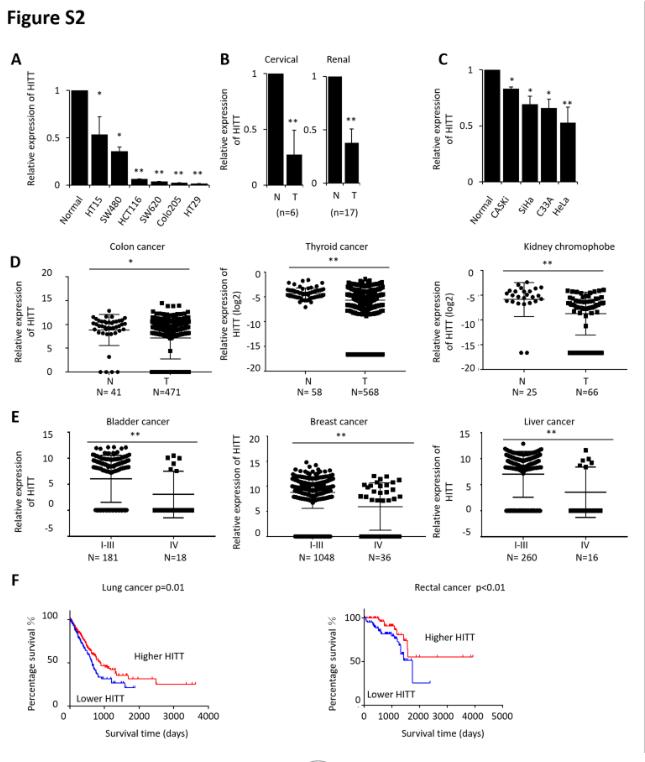
2、证明LncRNA HITT在人类癌症中普遍减少
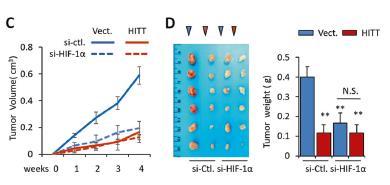
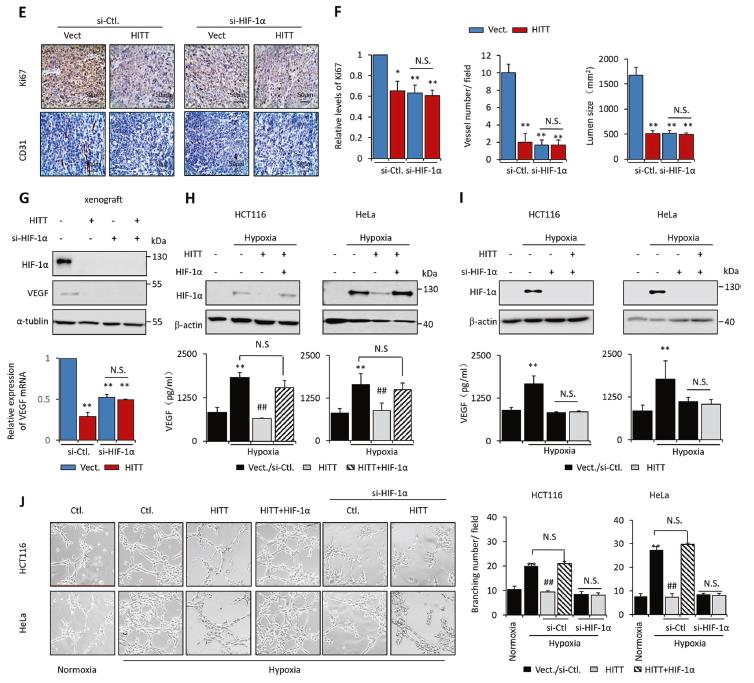
3、HITT以HIF-1α依赖性方式抑制血管生成和肿瘤生长
4、验证HITT抑制HIF-1α翻译且HITT充当HIF-1α翻译调节YB-1的诱饵
5、HIF-1α通过MiR-205介导的RNA降解
6、HIF-1α促进HITT的降解并通过反馈回路降低了HITT的稳定性
7、HITT下调与人类结肠癌组织中高水平的HIF-1α相关
研究结果:
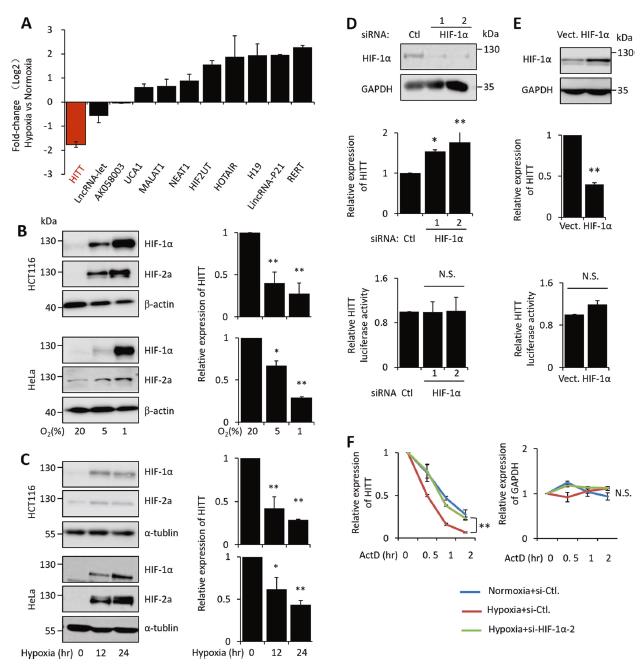
1、LncRNA HITT通常在人类癌症中减少
快速扩增3'-和5'-互补DNA末端分析证实HITT是具有聚腺苷酸化的20-50nt转录本位点。 HITT的编码电势值为0.387,在细胞核和细胞质中都检测到了可比的HITT量。值得注意的是,HITT在分析的结肠癌组织中显着下调。TCGA数据库的分析显示,HITT在多种癌症类型中被下调。HITT通过以HIF-1α依赖性方式抑制血管生成来防止体内肿瘤生长。在HCT116异种移植物中分别通过WB和qRT-PCR测量HIF-1α和VEGF蛋白和VEGF mRNA水平。这些发现共同表明HITT在人类癌症中通常被下调。
2、HITT以HIF-1α依赖性方式抑制血管生成和肿瘤生长
HITT可能通过拮抗有害的微环境因素来抑制肿瘤的生长。使用特异性靶向HIF-1α的siRNA抑制了HIF-1α的表达。HITT表达或HIF-1αKD降低了Ki-67的强度和血管密度。VEGF是已知的HIF-1α下游促血管生成靶标被HITT抑制。使用体外模型验证了HITT对HIF-1α诱导的血管生成的影响。HIF-1αKD还消除了低氧诱导的VEGF表达和HUVEC分支,有趣的是,完全消除了HITT的抗血管生成作用。相反,HIF-1α的过表达可以挽救HITT抑制的HUVEC分支。总的来说,该发现表明HITT通过干扰HIF-1α依赖性信号来抑制血管生成和肿瘤生长。
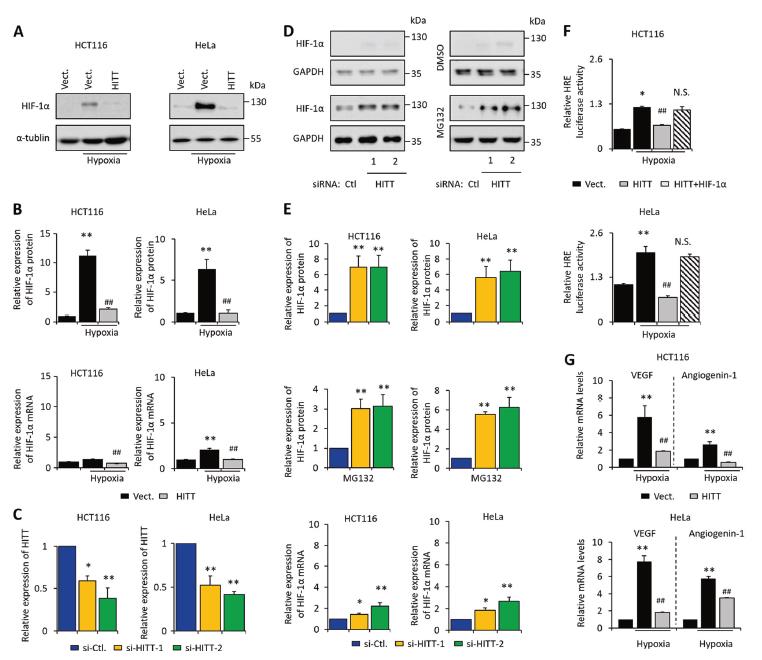
3、HITT抑制HIF-1α的表达和活性
HITT明显降低了缺氧诱导的HIF-1α表达约5至6倍,而HITT KD在常氧下增加了HIF-1α。HITT持续抑制HIF-1αmRNA表达约两倍,而HITT KD促进它的表达。表明HITT主要在蛋白质水平上调节HIF-1α并且HITT对HIF-1α的抑制作用可能是特异性的。更重要的是,缺氧显着增加了HIF-1α的转录活性,并且在稳定的HITT表达细胞系中这种作用被消除,而异位HIF-1α表达抑制了HITT的作用。另外,低氧增加了成熟的HIF-1α靶标的表达,例如VEGF和血管生成素-1,而HITT显着减弱了它的表达。这些数据共同说明,HITT主要通过调节其HIF-1α的蛋白水平来抑制其转录活性。
4、HITT抑制HIF-1α翻译
在HITT转染的稳定系和对照系中用蛋白合成抑制剂CHX处理细胞后,HIF-1α以相似的速率衰减。与多核糖体相关的HIF-1αmRNA随缺氧而增加,重要的是,异位HITT表达损害了缺氧诱导的HIF-1αmRNA与多核糖体的结合,而HITT KD增加了它们的结合。相反,GAPDH mRNA的分布均保持不变。HITT的过表达抑制了新合成的HIF-1α蛋白,而HITT KD在缺氧条件下使其表达增加。新合成的α-微管蛋白用作阴性对照,并且未被异位HITT表达或KD改变。因此,HITT抑制HIF-1αmRNA的翻译。
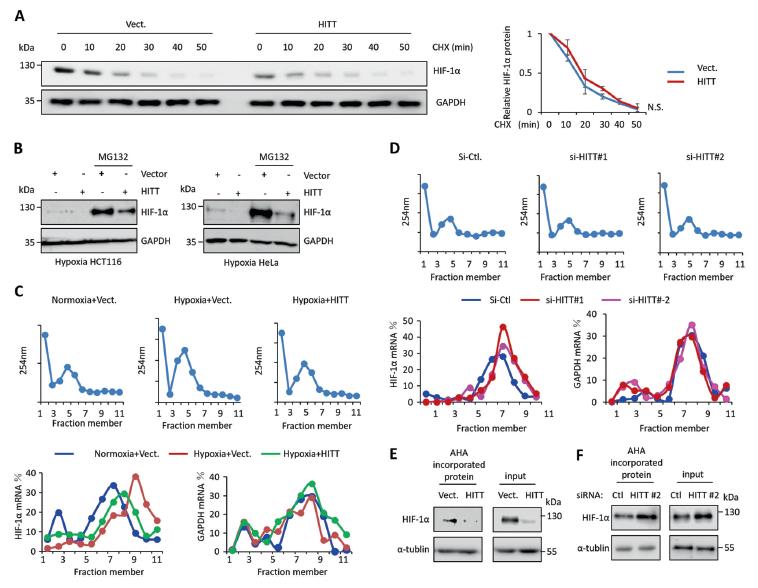
5、HITT充当HIF-1α翻译调节YB-1的诱饵
HITT对HCT116裸鼠异种移植物的体外和体内YB-1蛋白表达均无明显影响,但HITT中预测了8个YB-1结合序列。 YB-1 KD降低了HIF-1α水平,而HITT失去了影响YB-1 KD细胞中HIF-1α表达的能力。HITT的富集显着降低,而5'-UTR的富集响应于低氧开关而增加。在缺氧条件下并同时废除了HIF-1α 5'-UTR与YB-1的缺氧促进的关联。含有预测的YB-1结合基序的HITT有助于其与YB-1结合。表明HITT介导的HIF-1α抑制需要YB-1的结合。此外,YB-1 KD和异位HITT表达均阻止了低氧诱导的WTHIF-1α-5'-UTR-萤光素酶报道基因的活性,说明HITT直接与YB-1结合并阻止其与HIF-1α5'-UTR缔合,从而导致HIF-1α翻译减少。
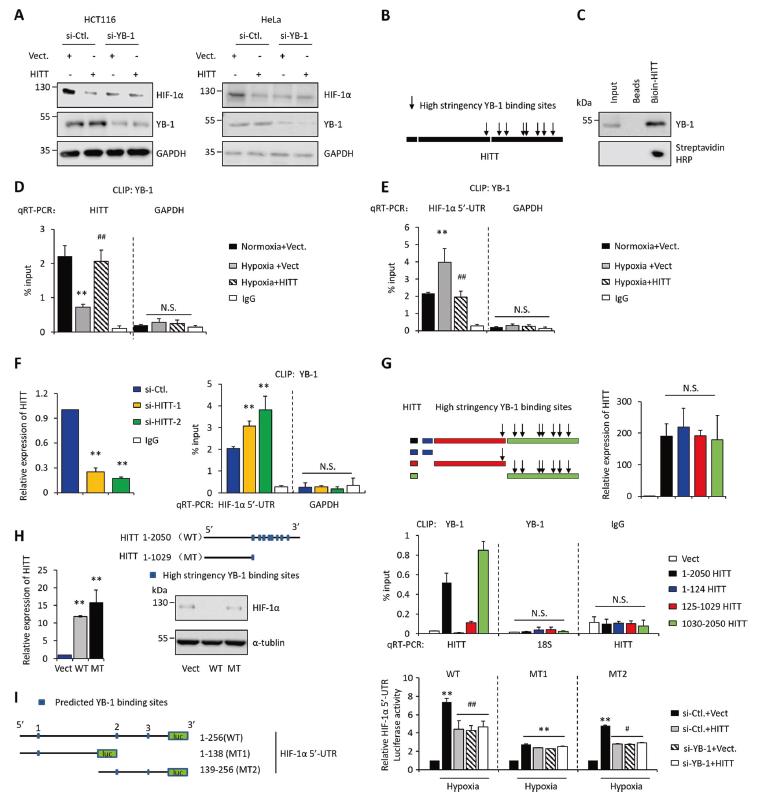
6、HIF-1α通过反馈回路降低了HITT的稳定性
在低氧条件下,HITT显着降低。si-HIF-1α降低了HIF-1α的水平,同时恢复了HITT的表达。相反,异位HIF-1α表达降低了HITT。HIF-2α对HITT的表达没有任何明显的影响,表明HITT受HIF-1α通过反馈回路的调控。在HITT的启动子中没有发现HRE基序,异位HIF-1α的表达和KD都不会对HITT启动子驱动的荧光素酶活性产生显着影响。缺氧会降低HITT的半衰期,而不降低GAPDH mRNA的半衰期。HIF-1αKD阻断了缺氧的影响。在缺氧和缺氧的情况下,HIF-1α缺乏的786-O细胞中检测到的HITT水平相当。这些数据表明,HIF-1α决定了HITT的稳定性。
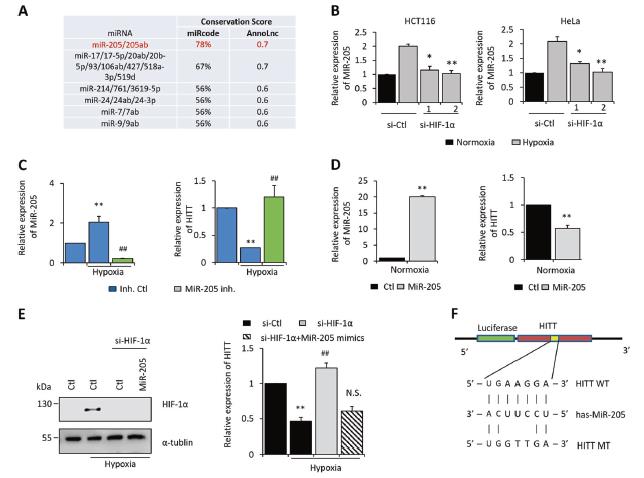
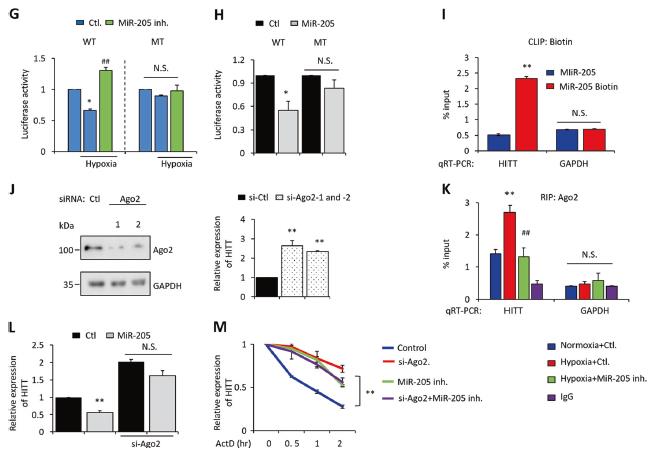
7、HIF-1α通过MiR-205介导的RNA降解促进HITT的降解
HITT是MiR-205的候选靶标。通过qRT-PCR验证MiR-205可能介导HIF-1α诱导的下游分子,MiR-205抑制剂预防了低氧诱导的MiR-205,并且还恢复了低氧条件下降低的HITT水平,而在常氧条件下通过转染MiR-205模拟物诱导的MiR-205显着降低了HITT。 MiR-205模拟物还消除了缺氧条件下HIF-1αKD诱导的HITT恢复。此外,缺氧降低了WT HITT荧光素酶报道基因的活性。通过下拉生物素化的前体MiR-205和qRT-PCR,证实了MiR-205和HITT之间的直接缔合。Ago2的KD是RNA诱导的沉默复合物/ MiR-205的重要组成部分。 RIP Ago2显示HITT与Ago2相关。这些数据构建了一个模型, Ago2 / miR-205在生理上与HITT结合,从而促进了MiR-205介导的HITT降解。
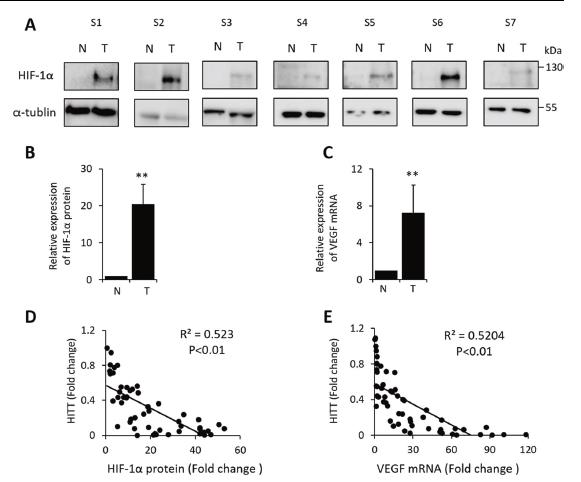
8、HITT下调与人类结肠癌组织中高水平的HIF-1α相关
发现了新颖的HITT–HIF-1α轴后,验证了HIF-1α与HITT在人类癌症组织中的表达之间的相关性。 在人结肠癌组织和匹配的对照中检测了HIF-1α及其靶标VEGF的蛋白水平(n = 46)。与匹配的正常对照相比,结肠癌组织中的HIF-1α和VEGF蛋白水平通常升高。降低的HITT水平与增加的HIF-1α蛋白和VEGF mRNA表达密切相关。这些发现与体外和异种移植数据相结合,证明HITT在翻译水平上调节HIF-1α,这导致HIF-1α及其靶标的表达降低。
总结:
癌细胞中HITT表达的恢复以HIF-1α依赖性方式抑制体内的血管生成和肿瘤生长。研究表明HITT主要通过干扰其翻译来抑制HIF-1α的表达。HITT通过高严格的YB-1结合基序从HIF-1αmRNA的5'- UTR滴定YB-1。HITT和HIF-1α表达之间的反向相关性在人结肠癌组织中得到进一步证实。此外,缺氧诱导的HIF-1α表达需要HITT下调。进一步证明HITT和HIF-1α形成了一个自动调节反馈回路,其中HIF-1α通过诱导直接靶向HITT降解的MiR-205破坏了HITT的稳定性。突出了HITT–HIF-1α轴构成了另一层血管生成和肿瘤生长的调节层,对治疗靶向具有潜在的影响。
综上所述,我们已经鉴定出HITT,一种新型的lncRNA。我们的数据强调了新发现的HITT–HIF-1α轴在调节血管生成和肿瘤生长中的重要性。HITT介导的HIF-1α合成的分子机制的详细剖析提供了通过恢复HITT活性抑制HIF-1α的新策略的见解。