






p53抑制肿瘤发生的奥秘
肿瘤抑制因子p53转录激活靶基因,在应激状态下抑制细胞增殖。p53也与原癌基因Myc的抑制有关,但机制尚不清楚。因此,小编和大家分享一篇近期发表于molecular cell上的文章“p53 Activates the Long Noncoding RNA Pvt1b to Inhibit Myc and Suppress Tumorigenesis”。在这里,我们确定了Pvt1b,一种lncRNA Pvt1的p53依赖性亚型,表达了Myc下游的50kb,它是由DNA损伤或致癌信号引起的,并在其转录位点附近积累。我们研究表明,在不改变基因座染色质组织的情况下,Pvt1b RNA的产生对于抑制顺式Myc转录是必要且充分的。抑制Pvt1b增加Myc水平和转录活性,促进细胞增殖。此外,在肺癌小鼠模型中,Pvt1b缺失加速了肿瘤生长。这些研究结果表明,Pvt1b在p53和Myc转录网络的交叉点发挥作用,增强p53的抗增殖活性。

结果:
1.p53在基因毒性和致癌应激条件下抑制Myc
为了深入了解p53抑制Myc的机制,我们模拟p53依赖的应激反应。为了模拟细胞对基因毒性应激的反应,我们使用野生型小鼠胚胎成纤维细胞(MEFs),用基因毒性药物Doxo处理(图1A)。我们观察到,在Doxo处理24小时后,p53转录程序的激活导致p21的3倍诱导,同时Myc RNA和蛋白水平降低(图1B,C)。我们还发现,在小鼠肺腺癌细胞系中,p53激活是由肿瘤基因应激,以他莫西芬-CreER依赖性恢复内源性p53表达为模型的,同样导致p21的激活和Myc RNA和蛋白质表达的下降(图1D-F)。在暴露于辐照6 Gy的肠道上皮细胞中,Myc抑制率为39%±5%(图1G-H)。接着为了阐明p53激活导致Myc抑制的机制,我们检查了p53是否与Myc位点相关。我们观察到,在Doxo处理的MEFs和tamo处理的KPR细胞中,应力依赖性的Myc抑制伴随着p53与位于Myc下游50 kb处的远端p53RE的结合(图1I)。与p53依赖相一致,p53存在的MEFs中,Myc RNA和蛋白水平的变化是存在的(图1J,1K)。此外,Myc RNA水平的降低在p53激活4h后可以检测到,与Myc蛋白水平的减少相吻合(图1L和1M)。使用Cycloheximide抑制蛋白翻译表明,压力的存在并未显著影响Myc蛋白的稳定性(图1N)。
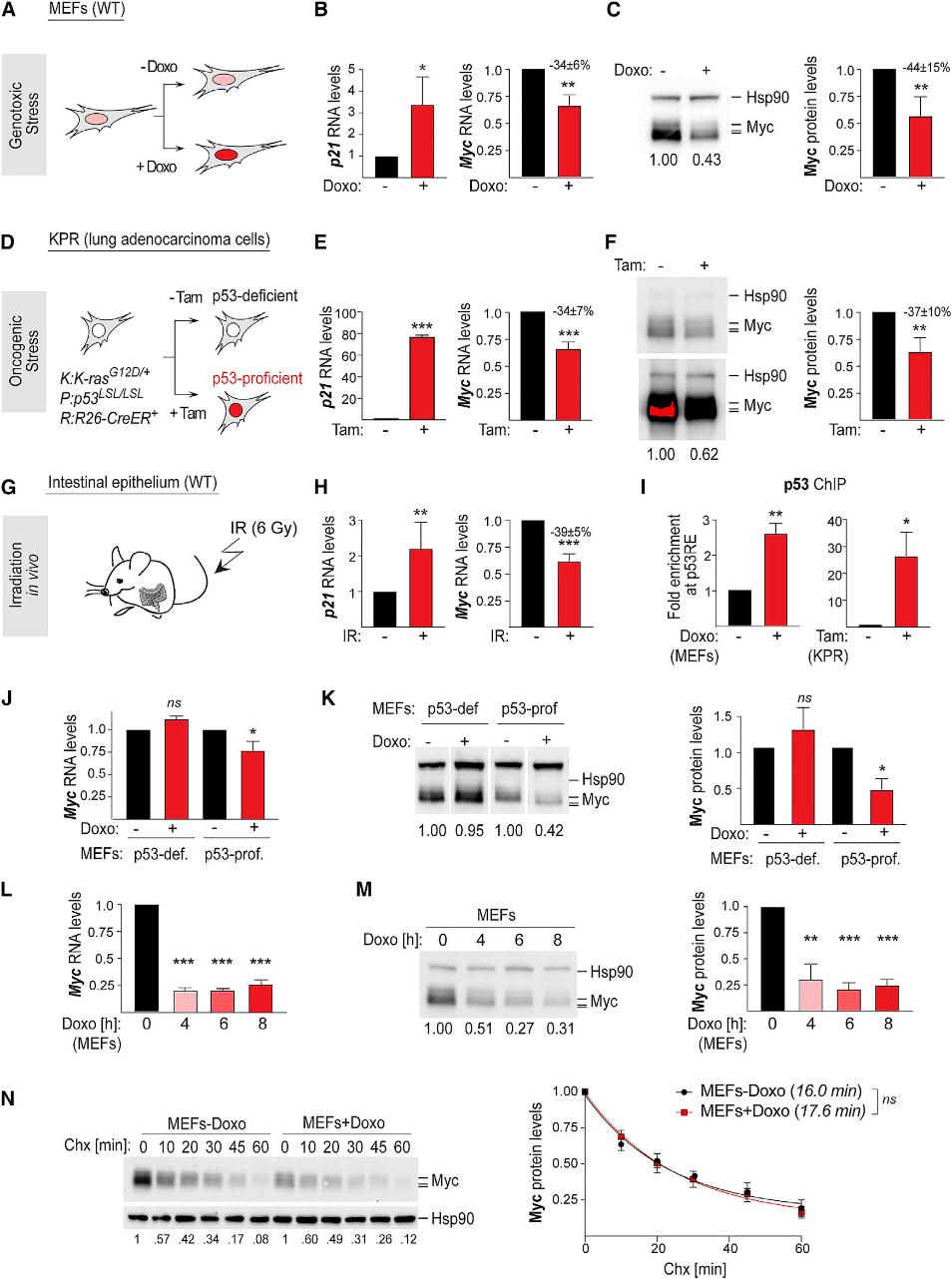
2.Myc抑制与Pvt1b的激活相关
考虑到lncRNAs可以顺式调控邻近基因的转录,我们检测了Pvt1是否在胁迫时起到了限制Myc表达的作用(图2A)。我们观察到经Doxo处理的MEFs中Pvt1b的诱导率为3.1±0.2倍(图2B),经Tam处理的KPR细胞中Pvt1b的诱导率为38±6倍(图2C)。为了进一步鉴定Pvt1位点产生的转录本,我们使用位于1a或1b外显子的正向引物和位于5外显子的反向引物进行RT-PCR。我们发现了广泛的选择性剪接的证据,证实了含有外显子1b的突变是由p53诱导的,而含有外显子1a的突变是组成性表达的(图2D和2E)。尽管存在剪接异质性,但初期RNA测序显示,仅通过使用外显子1b和外显子1a,应力诱导的Pvt1b与本构性表达的Pvt1a有所不同,并表现出与下游外显子类似的剪接模式(图2F)。
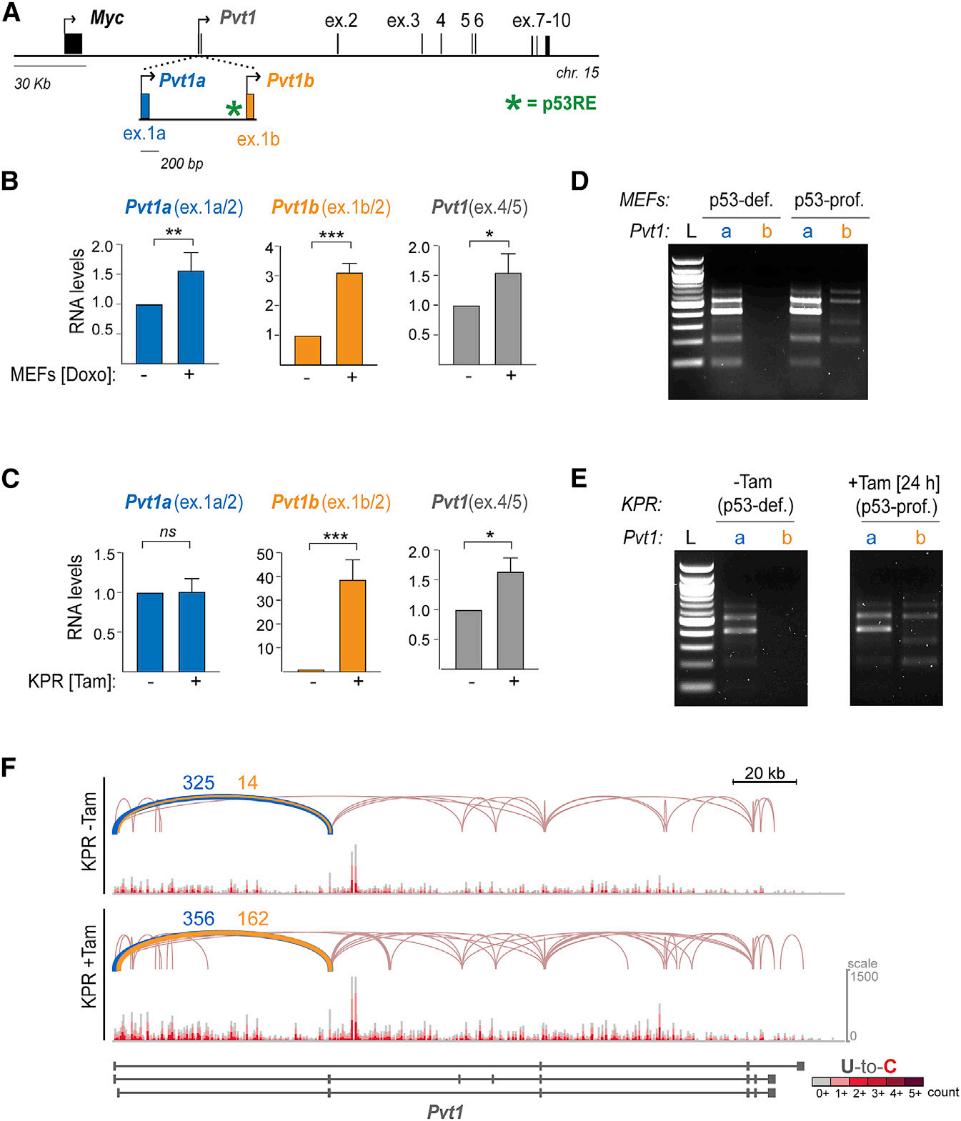
3.Pvt1b在Pvt1-Myc位点周围的染色质中积累
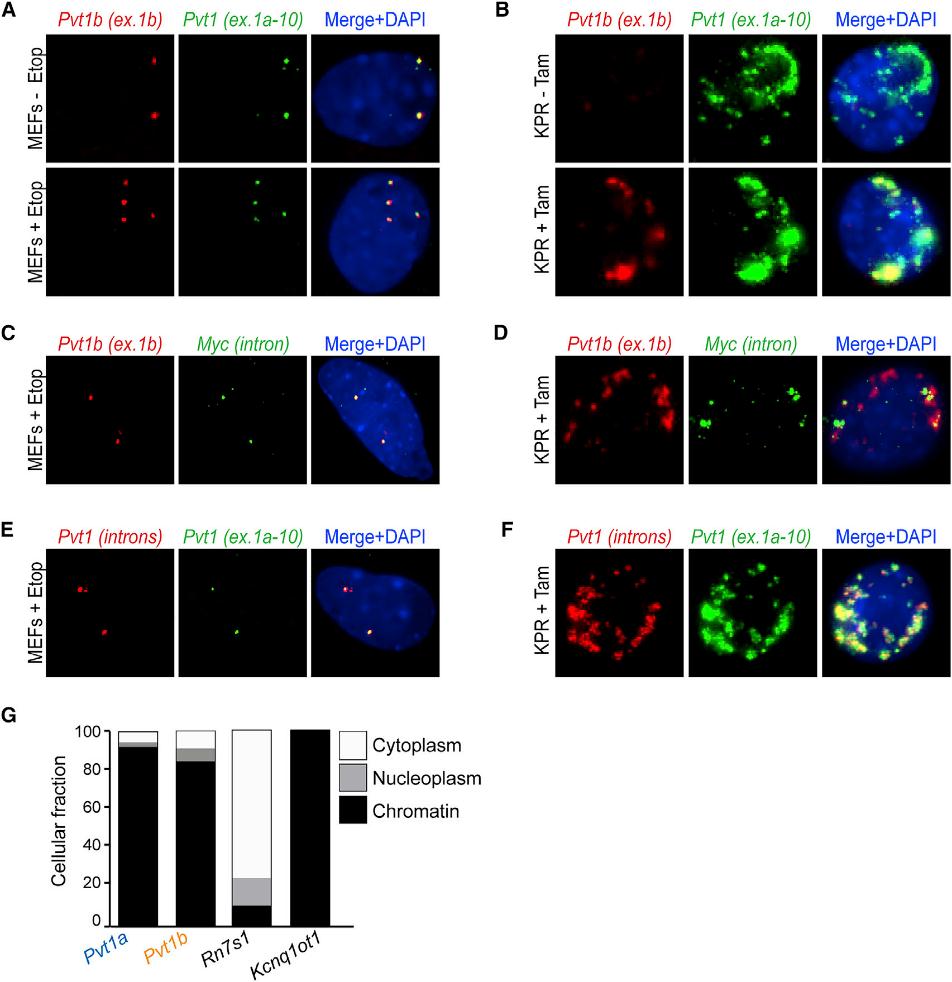
为了深入了解Pvt1b的潜在调控功能,我们进行了单分子RNA荧光原位杂交。我们观察到Pvt1a和Pvt1b在依托泊苷(Etop)处理的MEFs中主要表现为2点或4点核模式,分别反映了细胞周期的G1期或S/ G2期(图3A)。Pvt1a和Pvt1b在经Tam处理的KPR细胞中形成较大的云团(图3B)。值得注意的是,含有Pvt1a和Pvt1b的位点与新生Myc内含子特异性信号共定位,也与新生的Pvt1转录本共定位(图3C-3F)。这些结果使我们得出结论,转录后,Pvt1a和Pvt1b保留在围绕Pvt1-Myc位点的染色质上。亚细胞分馏分析证实了这两种Pvt1变体在染色质组分中均有富集(图3G)。
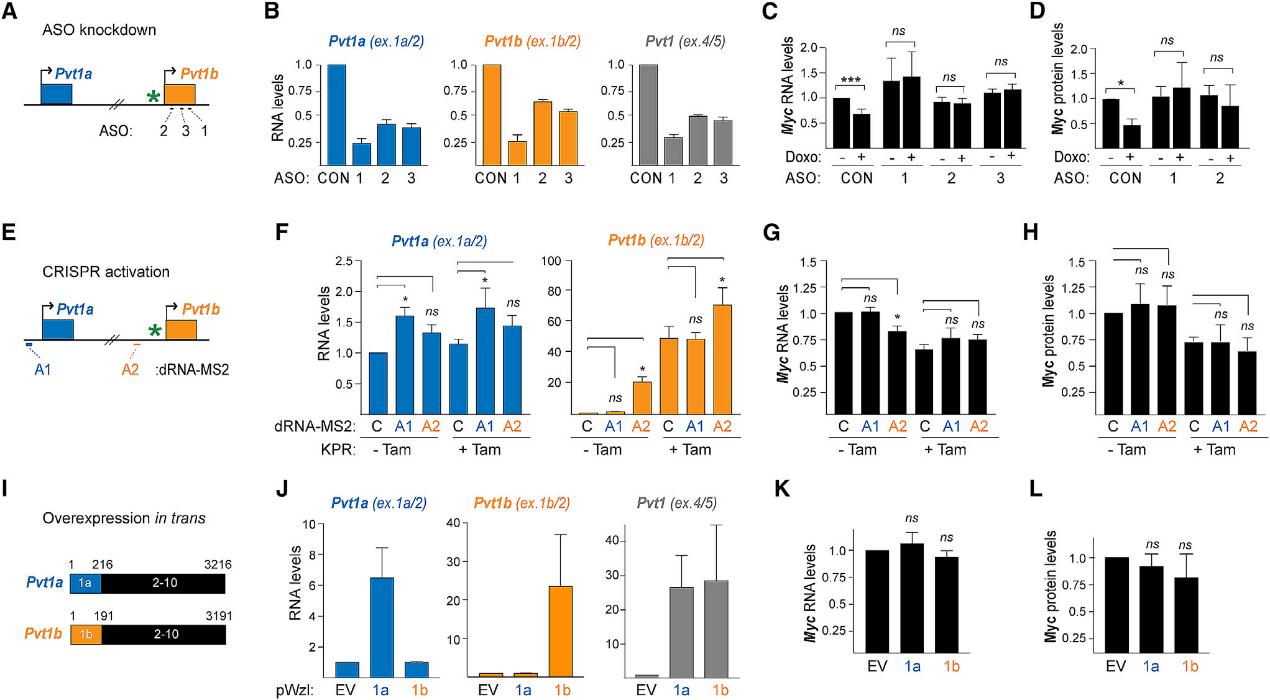
4.Pvt1b RNA抑制了Myc水平
基于Pvt1b的应力依赖性表达及其局部染色质积累,我们推测Pvt1b可能通过RNA依赖性机制参与Myc的抑制。为了直接验证这一假设,我们设计了三个针对1b外显子的独立反义寡核苷酸(ASOs)(图4A)。我们使用无目标的ASO作为阴性对照。由于ASOs会导致共转录RNA的裂解和降解,ASO1、ASO2和ASO3会显著下调Pvt1a和Pvt1b(图4B)。接下来,我们研究了Pvt1靶向ASOs对Myc表达水平的影响。在未处理的MEFs中,与CON样本相比,在ASO病例中,Myc RNA和蛋白水平没有发生显著变化, (图4C,4D)。与预期一样,使用Doxo治疗后,对照的MEFs的Myc RNA和蛋白水平显著下降。另一方面,我们发现以pvt1为靶点的ASOs完全挽救了应力诱导的Myc RNA和蛋白的下调。这些发现表明,p53对Pvt1b的转录激活是Myc在应激状态下抑制的必要条件。为了检测Pvt1b是否能充分抑制Myc,我们使用CRISPR-SAM系统激活p53缺陷细胞内源性Pvt1b的表达。我们分别设计了针对Pvt1a和Pvt1b启动子的A1和A2 dRNA-MS2(图4E)。与非靶向对照相比,使用A1的CRISPRa在不改变Pvt1b水平的情况下诱导了1.6倍的Pvt1a,而使用A2导致了20倍的Pvt1b活化(图4F)。接下来,我们检测了内源性Pvt1a和Pvt1b的激活对Myc水平的影响。p53修复后,Pvt1b的激活并没有进一步下调Myc水平,这说明Pvt1b的作用是在p53的下游(图4G)。另一方面,Pvt1b的激活不足以抑制Myc蛋白水平,这为在转录后水平独立输入Pvt1b提供了可能(4H)。然后,为了区分顺式和反式的活性,我们通过转染含有外显子1a-10(1a)或1b-10(1b)的cDNA构建物来检测外源性的Pvt1a和Pvt1b是否会影响Myc的表达(图4I)。我们观察到Pvt1a的过表达为6.5倍,而Pvt1b的过表达为23倍,与CRISPRa诱导的过表达相当(图4J)。然而,我们发现外源运输的Pvt1a或Pvt1b对Myc RNA或蛋白水平没有显著影响,这与反式运输的影响相反(图4K、4L)。
5.Pvt1b的遗传抑制逆转应力诱导的Myc下调
为了研究Pvt1b对p53抑癌途径的功能贡献,我们开发了一种基因方法,通过改变表达所需的p53RE来特异性抑制Pvt1b的表达。我们通过Sanger测序证实了p53RE的突变,并通过染色质免疫沉淀证实了∆RE突变将p53的结合降低了15倍(图5A,5B)。通过qRT-PCR,与对照组相比,∆RE细胞中的Pvt1b水平被显著抑制(图5C),而通过smRNA-FISH,我们观察到经Tam处理的∆RE KPR细胞中Pvt1b特异性信号的丢失(图5D,5E)。这些观察使我们得出结论,Pvt1b相关的p53RE突变导致了应力依赖性Pvt1b活化的有效消除。接下来,我们通过qRT-PCR和免疫印迹法检测了∆RE突变以及由此导致的Pvt1b表达缺失对细胞应激反应中Myc水平的影响。在Con KPR群体、KPR克隆和MEF系中,暴露于致癌或基因毒性应激导致的Myc RNA和蛋白水平预期显著下降(图5F-H)。与非应激细胞相比,∆RE KPR群体、KPR克隆和MEF系暴露于应激并不导致Myc RNA水平显著下降,这与ASO数据一致。这些结果为Pvt1b调控p53下游的Myc RNA水平提供了独立的遗传验证。
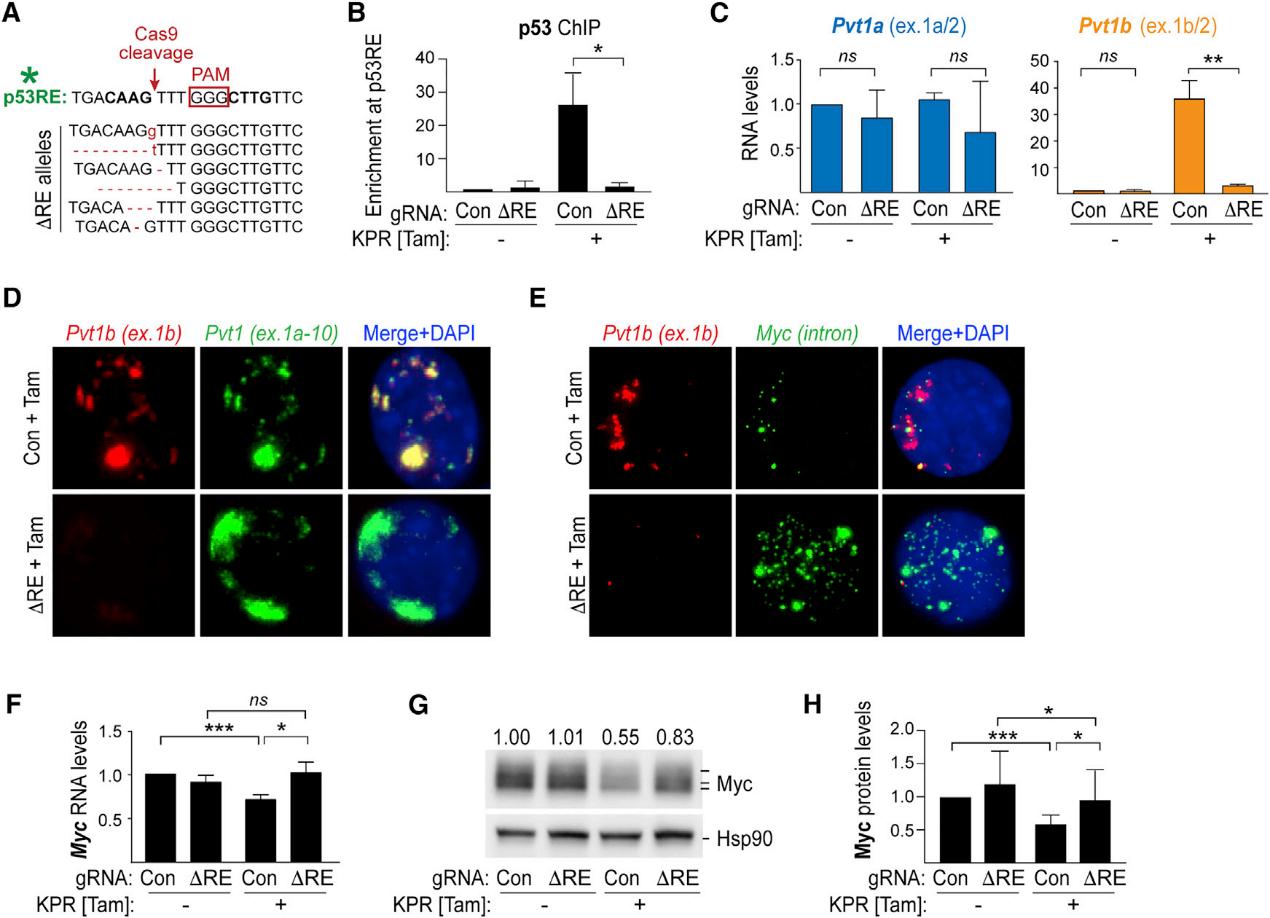
6.Pvt1b抑制了Myc转录活性和细胞体外增殖
通过分析∆RE突变对未处理、Doxo处理的∆RE和Con MEFs的总RNA的基因表达的影响,我们证实Myc是Pvt1b应对应激的调控靶点(图6A)。接下来,为了测试Pvt1b是否在转录水平或转录后水平发挥作用,我们对未处理和经Tam处理的∆RE和Con KPR细胞新生RNA进行了测序。我们发现,与Con+Tam KPR细胞相比,∆RE+Tam细胞中新生的Myc转录本显著上调(图6B,6C)。这些数据表明,Pvt1b的产生促进了Myc的转录抑制。接下来,我们通过检测196个Myc靶基因中Pvt1b缺失的后果,探讨Myc RNA水平的变化如何影响Myc转录程序。与随机产生的一组表达水平相当的对照基因相比,我们发现MEFs和KPR细胞中Myc靶点的水平显著升高(图6D,6E)。考虑到Myc靶基因包含促进细胞生长的因子,我们将突变细胞的增殖与对照组进行了比较。与对照组相比,Pvt1b表达的丢失导致细胞增殖和集落形成显著增加(图6F,6G)。这些数据表明,Pvt1b通过介导p53功能的特异性来抑制细胞的体外增殖。
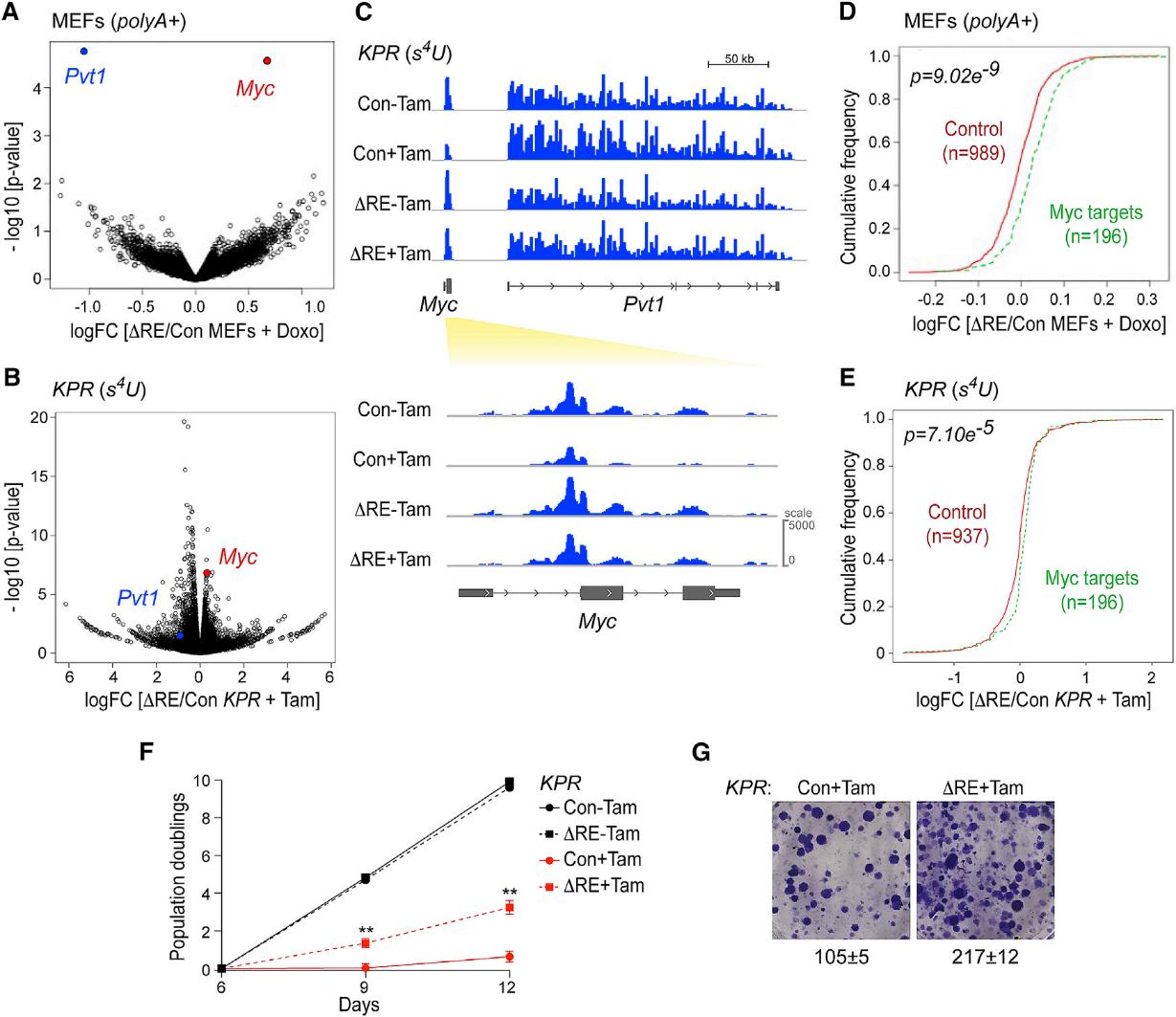
7.Pvt1b的肿瘤特异性抑制促进了体内的肿瘤生长
为了阐明Pvt1b是否在体内介导p53功能的某些方面,我们对Pvt1b相关的p53RE进行了肿瘤特异性诱变(图7A)。Sanger测序证实了在UGPC-∆RE感染动物中成功突变了pvt1b相关的p53RE(图7B)。接下来,我们检查了感染UGPC-Con、UGPC-p53KO和UGPC-∆RE病毒的小鼠肺部的HE切片,并在肿瘤发生16周后处死。组织病理学分析显示,在感染UGPC-Con的动物中,所有的肿瘤都表现出了1级特征。相比之下,70%的表达UGPC-p53KO的肿瘤以非典型核、间质增生和向低分化表型转化为特征,并被划分为2级或3级(图7C,7D)。另一方面,肿瘤面积相对于总肺面积的定量显示,UGPC-∆RE小鼠的肿瘤负荷较对照组小鼠显著增加(图7 E)。值得注意的是,p53RE编辑的小鼠的肿瘤负荷与UGPC-p53KO小鼠的肿瘤负荷相当(图7E)。这些发现表明,Pvt1b在很大程度上介导了p53下游的生长抑制功能,尤其是在疾病的癌前病变阶段。与UGPC-Con小鼠相比,感染UGPC-∆RE的动物的肿瘤负荷增加并不是由于细胞凋亡减少。相反,肿瘤负荷的增加可能是由于增殖的增强。在Pvt1b缺陷肿瘤中,来自UGPC-∆RE感染动物的肿瘤的磷酸化组蛋白H3 阳性的有丝分裂细胞明显多于来自UGPC-Con感染小鼠的肿瘤(图7F,7G)。最后,为了研究Pvt1b是下游作用还是独立于p53,我们进行了上位实验。我们分析了UGPC-Con或UGPC-∆RE病毒在肿瘤发生12周后的肿瘤负荷。与上述结果一致,我们观察到UGPC-∆RE感染小鼠与UGPC-Con感染的KC动物相比,肿瘤负荷显著增加。相反,我们发现感染UGPC-∆RE和UGPC-Con的KPC动物的肿瘤负荷没有显著差异。感染UGPC-∆RE的KC小鼠与感染UGPC-Con的KPC小鼠的肿瘤负荷差异无统计学意义(图7H)。这些结果表明,Pvt1b和p53通过一个共同 的途径增强了癌前肿瘤的扩散。
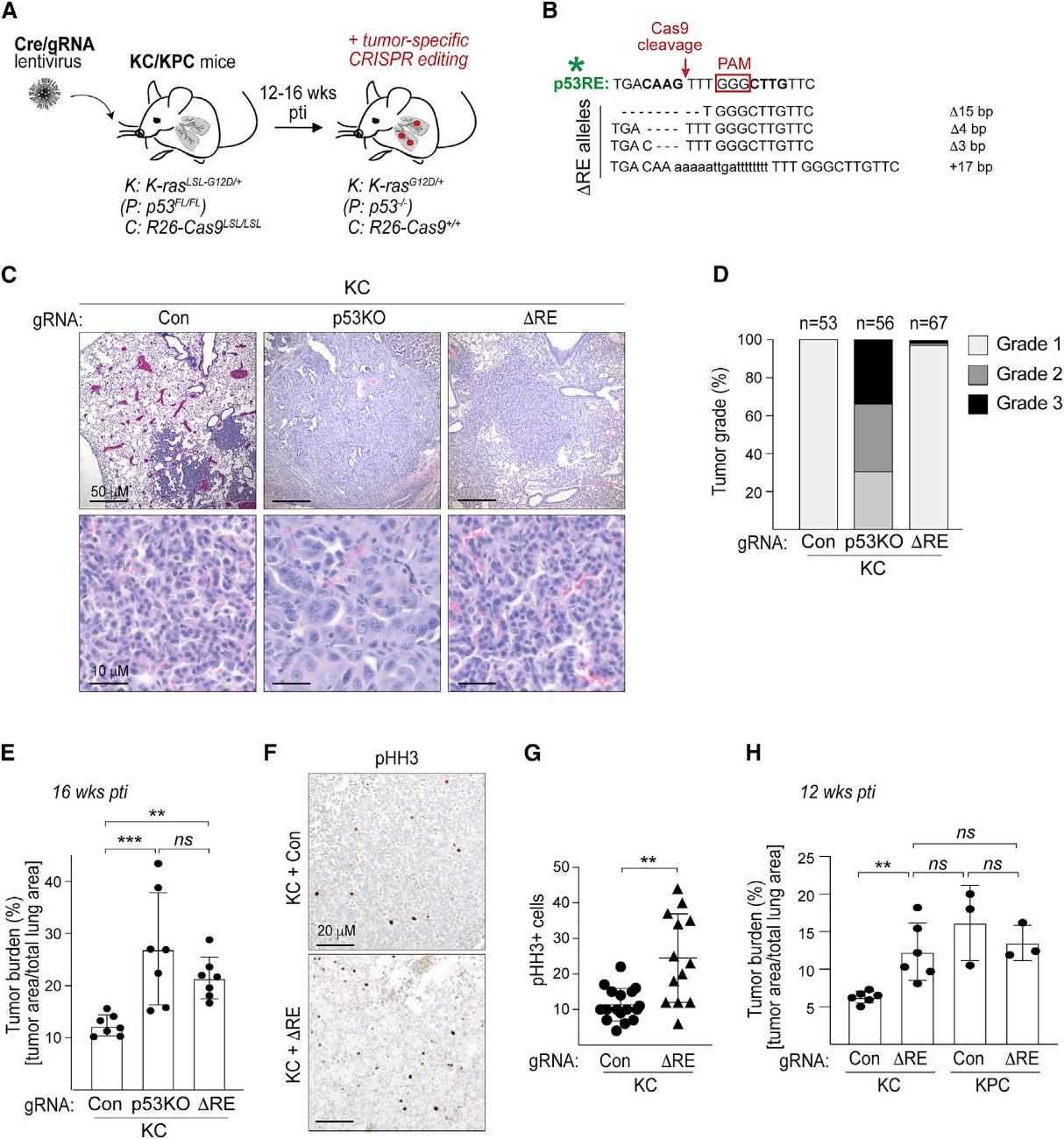
结论:
保守的lncRNA亚型Pvt1b是一种位点特异性的转录调节因子,在p53介导的应激反应中抑制Myc的转录。Pvt1b RNA的产生抑制细胞增殖和肿瘤生长,揭示了这种癌症相关的lncRNA的肿瘤抑制活性。
