






自噬与血管重塑
跨膜成员16A(TMEM16A)是钙激活的氯离子通道的一个组成部分,调节血管平滑肌细胞(SMC)的增殖和重塑。自噬是真核生物中高度保守的细胞分解代谢过程,在血管平滑肌细胞中发挥着重要的生理功能。TMEM16A是否参与自噬介导的血管重塑尚不清楚。接下来小编为大家带来发表于“Theranostics”上的文章“TMEM16A ameliorates vascular remodeling by suppressing autophagy via inhibiting Bcl-2–p62 complex formation”。在本研究中,我们研究了自噬对血管内SMC的增殖和重构的贡献,并研究了TMEM16A是否参与了这些过程。

结 果:
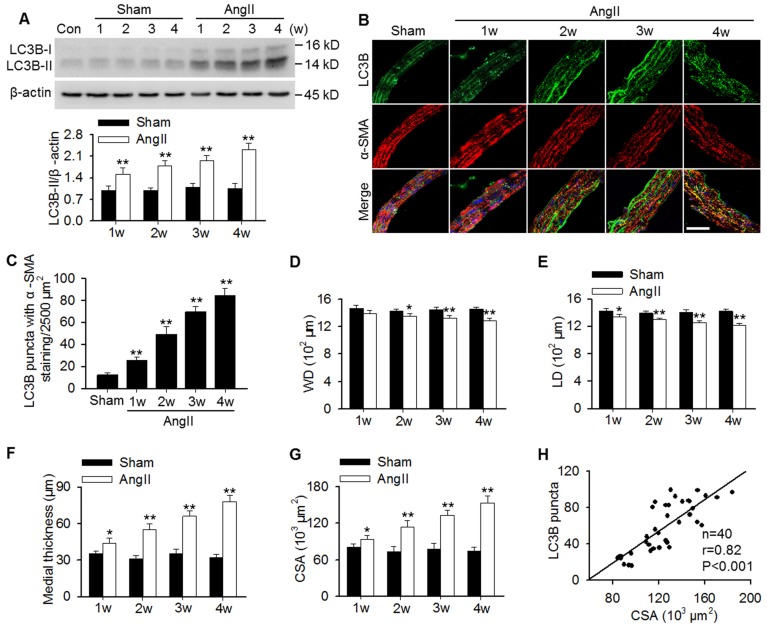
1.自噬水平与血管重塑呈正相关
为探讨自噬是否参与血管性高血压诱导的血管重塑,测定了自噬标志物LC3B的蛋白表达。结果显示,从术后1周开始,AngII诱导的高血压小鼠主动脉中LC3B-II的表达逐渐增加(图1A)。此外,AngII灌注显示主动脉平滑肌中LC3B阳性绿点的数量随时间而增加,如平滑肌标记物α-SMA(图1B和C)。尽管AngII引起的血管壁和管腔直径的减小是轻微的,但与相应的假手术组相比,差异是显著的(图1D和E)。因此,从术后1周开始,AngII灌注逐渐增加了中膜厚度和平均内侧CSA值(图1F和G)。值得注意的是,相关分析表明,LC3B点状结构与血管性高血压时胸主动脉内侧CSA值呈正相关(图1H),表明自噬可能在血管重塑中发挥作用。
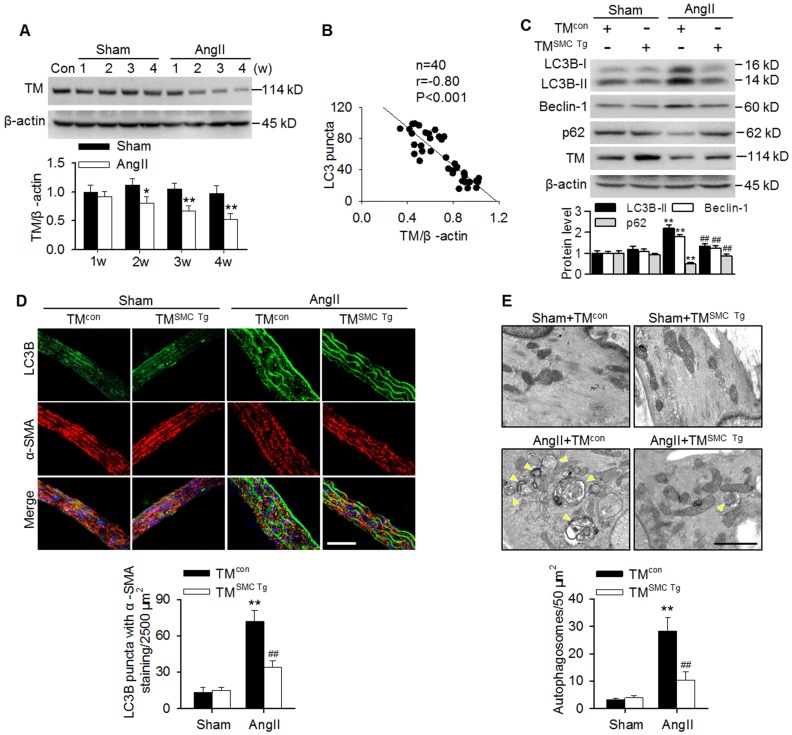
2.TMEM16A抑制大动脉血管中AngII诱导的自噬
与假手术小鼠相比,从术后2周开始,AngII高血压小鼠的主动脉中TMEM16A的表达逐渐减少(图2A)。有趣的是,TMEM16A表达的降低与AngII高血压小鼠主动脉中LC3B阳性点状数目呈负相关(图2B)。为探讨TMEM16A在高血压大鼠主动脉血管自噬中的作用,制备了TMEM16A-SMC特异性转基因小鼠(TMSMC-Tg),并注入AngII。如图2C所示,假手术TMcon和TMSMC-Tg小鼠主动脉中LC3B-II、Beclin-1和p62的表达差异不显著。然而,与TMcon小鼠相比,TMSMC-Tg小鼠主动脉血管内LC3B-II和Beclin-1表达的增加以及p62表达的减少均受到明显抑制。免疫荧光染色显示,SMC特异性TMEM16A的过度表达显著减弱了AngII诱导的增加的LC3B阳性点状细胞(图2D)。进一步的电镜观察显示,与假手术小鼠相比,AngII处理小鼠的胸主动脉中层自噬体增多。TMEM16A上调显著抑制自噬体的积累(图2E)。这些数据提示TMEM16A的表达在调节血管自噬中起重要作用。
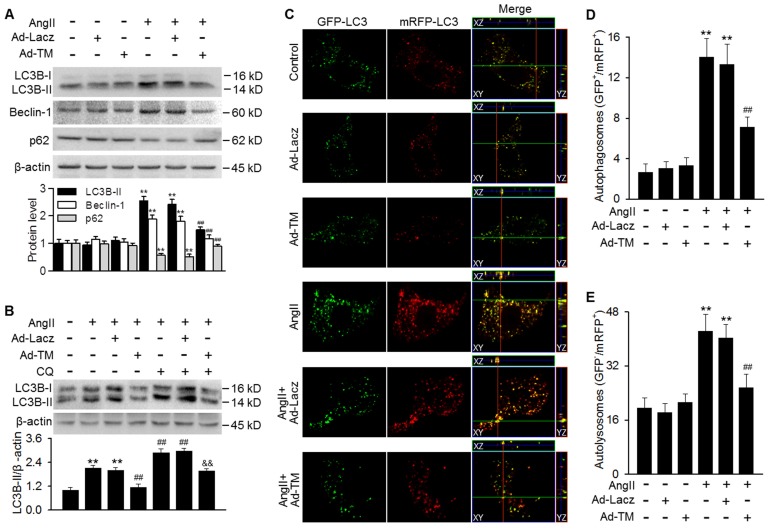
3.TMEM16A阻断AngII诱导的自噬通量
我们的研究结果显示,抑制TMSMC-Tg小鼠的AngII诱导的自噬,有助于我们研究TMEM16A在体外是否发挥类似的作用。结果表明,TMEM16A过表达显著抑制了AngII诱导的LC3B-II和Beclin-1表达的增加以及p62表达的减少(图3A)。为了区分LC3B-II和Beclin-1水平的升高是由于自噬体形成增加还是自噬体-溶酶体融合受损,我们研究了氯喹(溶酶体抑制剂)对LC3B-II表达的影响。结果表明,在抑制溶酶体活性后,经AngII处理的MASMCs中LC3B-II的表达进一步增强。然而,TMEM16A过表达显著逆转了氯喹对LC3B-II表达的影响,提示TMEM16A抑制了自噬小体的形成(图3B)。为了证实TMEM16A对自噬通量的影响,我们采用串联的mRFP-GFP-LC3腺病毒进行荧光分析。与mRFP相比,GFP荧光在较高pH值下是最佳的,并且在溶酶体酸性环境中可以更快地猝灭,这使得我们能够通过分别检测黄点(GFP+/mRFP+)和红点(GFP-/mRFP+)来监测自噬体和自溶体。经AngII处理的MASMCs可见丰富的黄色和红色斑点。然而,异位过表达TMEM16A与单独使用AngII治疗相比,显著减少了黄点和红点的数量(图3C-E),表明TMEM16A通过抑制自噬通量来减弱AngII诱导的自噬。
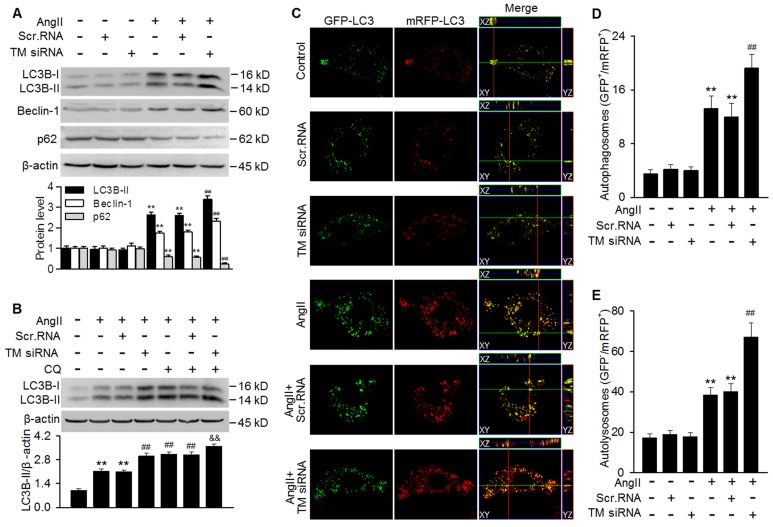
4.下调TMEM16A促进AngII诱导的自噬通量
为了进一步阐明TMEM16A在自噬调节中的作用,我们用siRNA敲除了TMEM16A在MASMCs中的表达。结果表明,TMEM16A基因敲除进一步增强了AngII诱导的LC3B-II和Beclin-1表达增加和p62表达减少(图4A)。此外,氯喹对经TMEM16A siRNA处理的细胞LC3B-II表达的影响增强(图4B),表明TMEM16A下调进一步促进自噬体成熟。与单用AngII处理相比,TMEM16A基因敲除显著增加了黄点和红点的数量(图4C-E),表明TMEM16A基因敲除增强了AngII诱导的自噬。
5.TMEM16A抑制AngII诱导的VPS34活性
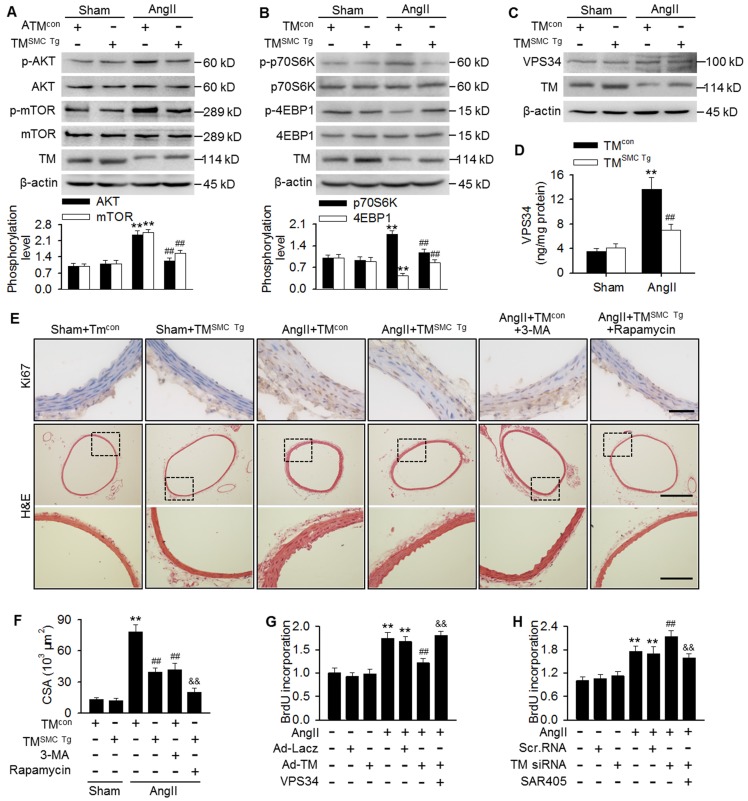
为了确定TMEM16A调节自噬的机制,我们研究了TMEM16A对mTOR途径的影响。出乎意料的是,与假手术小鼠相比,经AngII处理的小鼠主动脉中AKT、mTOR和下游靶点p70S6K的磷酸化显著增加,而4-EBP1磷酸化被抑制。相反,SMC特异性TMEM16A过表达显著抑制了mTOR途径的激活(图5A和B)。此外,AngII诱导的高血压TMSMC-Tg和TMcon小鼠主动脉中VPS34的表达相似(图5C)。AngII素输入显著增加了VPS34的激酶活性,而VPS34被SMC特异性TMEM16A过表达所抑制(图5D)。
考虑到TMEM16A和自噬在血管重塑中的作用,以及TMEM16A调节自噬的能力,我们探讨了自噬在TMEM16A介导的血管平滑肌细胞增殖和重塑中的作用。结果表明,在基础条件下,TMcon和TMSMC-Tg小鼠胸主动脉Ki67阳性细胞数和中间CSA值相似。然而,AngII注射显著增加了TMcon小鼠Ki67染色水平和CSA中间值,而这在TMSMC-Tg小鼠中受到抑制。此外,3-MA,一种靶向VPS34的自噬抑制剂,显著抑制AngII诱导的血管平滑肌细胞增殖和血管重塑(图5E和F)。值得注意的是,雷帕霉素,一种通过抑制mTOR信号传导的自噬刺激剂,并没有逆转TMEM16A上调对血管平滑肌细胞增殖和血管重塑的抑制作用,而是增强了这种作用(图5E和F)。此外,AngII诱导的MASMC增殖被TMEM16A过表达显著减弱,并被TMEM16A敲除增强(图5G和H)。VPS34的过表达消除了TMEM16A上调对AngII诱导的细胞增殖的抑制作用(图5G)。此外,用更高选择性抑制剂(SAR405)对VPS34的药理学抑制很大程度上消除了TMEM16A下调引起的细胞增殖增加(图5H)。总之,这些结果表明,抑制VPS34介导的自噬是TMEM16A对血管平滑肌细胞增殖和重塑作用的基础。
6.TMEM16A从p62/Bcl-2/Beclin-1/VPS43复合物中分离p62并抑制自噬的发生
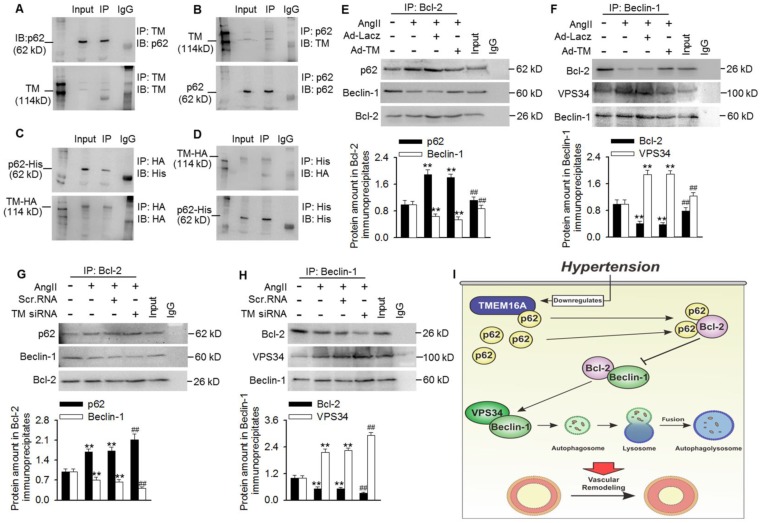
为了研究TMEM16A如何减弱VPS34的活性和随后的自噬通量,我们研究了TMEM16A在调节自噬起始中的作用。结果表明,TMEM16A与p62共免疫沉淀(图6A和B)。在转染HA-RFP-TMEM16A和His-p62的细胞中,TMEM16A始终与外源p62相互作用(图6C和D)。以前有报道称p62结合Bcl-2并阻止Bcl-2/Beclin-1复合物的形成。Bcl-2-Beclin-1相互作用的中断促进了Beclin-1-VPS34的关联,并增加了Beclin-1相关的VPS34活性。基于此,我们接下来检查这些相关蛋白质之间的关联。用AngII处理的MASMCs分别增加了p62-Bcl-2和VPS34-Beclin-1的结合,但减少了与Beclin-1共免疫沉淀的Bcl-2的数量(图6E-H)。TMEM16A过表达显著限制了p62-Bcl-2和VPS34-Beclin-1的形成,但逆转了Bcl-2-Beclin-1结合的减少(图6E和F)。相反,TMEM16A的抑制增强了p62-Bcl-2和VPS34-Beclin-1的结合,进一步破坏了Bcl-2-Beclin-1复合物的形成(图6G和H)。这表明TMEM16A至少部分地通过p62、Bcl-2、Beclin-1和VPS34之间的四向相互作用来调节VPS34的活性。
结 论:
总之,TMEM16A通过调节p62、Bcl-2、Beclin-1和VPS34之间的四向相互作用抑制自噬介导的VSMC增殖,降低VPS34的活性,从而改善血管重塑。这些发现为TMEM16A减轻血管重塑的机制提供了新的见解,并为该病的靶向治疗提供了新的思路。