






Nat. Methods | TooManyCells识别并可视化单细胞进化枝的关系
细胞转录输出有助于细胞类型和状态的确定。新兴技术,如单细胞RNA-seq (scRNA-seq)改善了识别和描述的细胞状态异质性由不同的基因表达程序和拥有先进我们理解细胞多样性的功能作用在细胞分化等领域,对刺激的回应,和失调疾病。
许多聚类算法建议对scRNA-seq数据进行划分,以识别具有相关转录程序的细胞群,将细胞划分为已知的细胞类型或状态,在某些情况下描述新的稀有细胞种群。基于louva的聚类方法s1是应用最广泛的scRNA-seq聚类方法之一。这些算法以贪婪的方式迭代地优化社区度量,即Newman-Girvan模块化,以生成单个的单元划分。在大多数scRNA-seq分析工作流程,确定细胞具有相似的转录组的集群模式然后使用降维可视化技术,如t-distributed随机邻居嵌入(t-SNE) 2,在高维基因表达空间随机减少到项目每个细胞的位置在一个二维曲面。这个工作流程导致识别单个单层的单元分区,这些单元使用缺乏定量的组间和组内关系信息的可视化方法进行解释。考虑到精确的细胞状态定义和国米和intra-relationships是上下文相关的,可能不适合一个uni-layer分区的细胞,而是一个层次结构的嵌套细胞状态,我们介绍了一个范式转变套件工具的分析,解释和单细胞的可视化数据通过启用同步描述和可视化的多层细胞演化支。
TooManyCells是一套基于图形的算法和工具,可在保持和呈现细胞群之间的相互关系的同时,高效,全局且无偏见地识别和鉴定细胞进化枝。TooManyCells提供了一种新颖的单细胞聚类算法ClusterTree,该算法为单细胞测量分析实现了有效的无矩阵分裂分层光谱聚类。此外,TooManyCells 能够评估细胞多样性并估计调查异质种群中特定水平的细胞物种丰富度所需的样本量。TooManyCells还提供BirchBeer了一个完全可定制的新颖模型,用于以单细胞分辨率可视化分层细胞聚类分析。在渲染聚类树时,BirchBeer利用着色标签,缩放分支,模块化叠加,内部节点标签,叶节点摘要等的加权平均混合,可以对单细胞进化枝进行多层和多方面的探索。为了提供最大的适用性,TooManyCells加载并生成几种标准文件格式。因此,可以将替代聚类算法与可视化结合使用,从而在多种方法之间提供协调,以帮助阐明单个单元格之间的关系。这些聚类和可视化算法能够描述依赖于上下文和应用程序的细胞集群,并加速探索和量化细胞分支之间的相互关系。
近日,来自宾夕法尼亚大学佩雷尔曼医学院的病理学和检验医学系,宾夕法尼亚大学佩雷尔曼医学院遗传学系,美国宾夕法尼亚州佩雷尔曼医学院的宾夕法尼亚大学艾布拉姆森家庭癌症研究所,法尼亚大学宾夕法尼亚大学表生遗传研究所课题组在Nat. Methods 发表题为TooManyCells identifies and visualizes relationships of single-cell clades的研究,报道了一种新软件——TooManyCells,可以识别并可视化单细胞进化枝的关系。
这组工具通过在依赖于上下文和应用程序的范围内同时比较单元状态,从而在分析和解释单细胞数据方面提供了范式转换。这种转变的一个副产品是对稀有种群的即时检测和可视化,其性能优于以前的算法,这是通过将这些工具应用于来自各种小鼠器官的现有单细胞RNA-seq数据集而得到证明的。
一、TooManyCells 用于单细胞分辨率的聚类和可视化
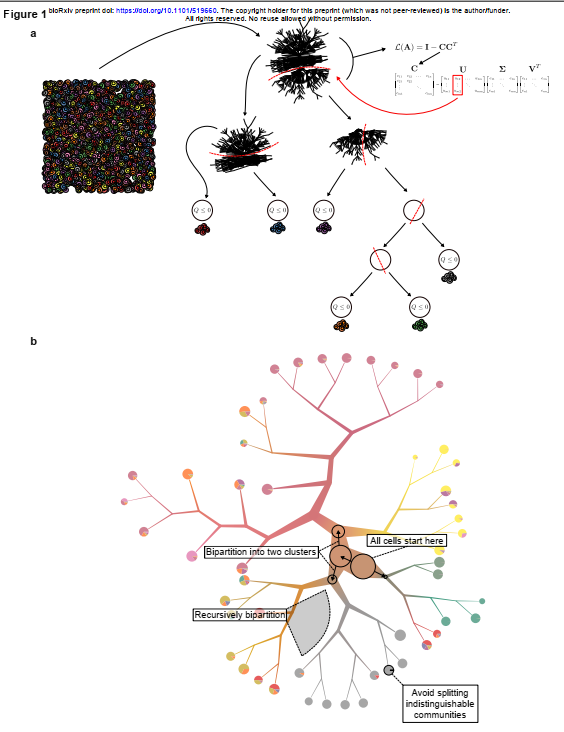
例如,通过scRNA-seq测量对递归结构进行递归划分可以概括脾脏中的淋巴样群体,在该群体中,分裂将B与T细胞分开,然后可以将血浆细胞谱系与过渡性B细胞分离,然后分离成浆细胞和浆细胞,直至达到具有不同B细胞受体结构的浆细胞的分离。要确定单元状态的连续体ClusterTree,TooManyCells,从属于单个组的所有单元格开始,作为聚类树的根(图1a和1b)。这些细胞由具有m个细胞和n个基因的m × n矩阵表示。矩阵中的每个条目都是细胞中某个基因的丰度数据。因此,ClusterTree生成嵌套单元簇的层次结构,其中每个内部节点都是给定规模的簇,叶节点是细粒度簇,其中任何附加的二分分割都将与单元的随机分裂一样好。
二、ClusterTree:用于scRNA-seq的无矩阵分裂分层光谱聚类算法
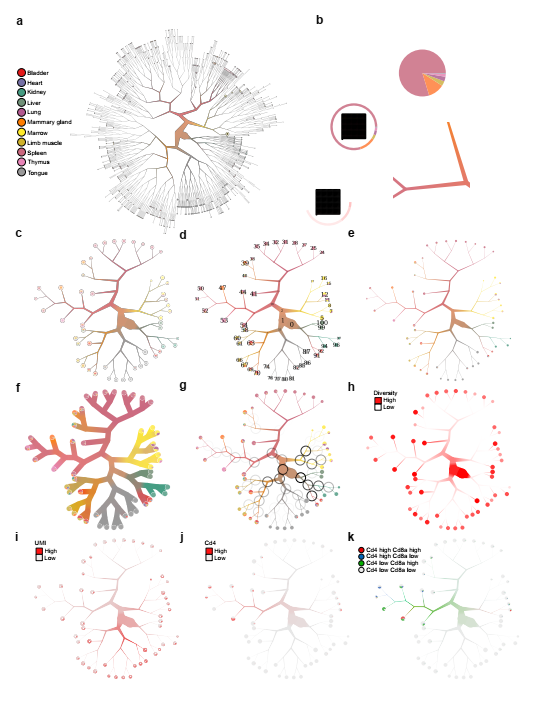
单细胞分辨率的可视化是scRNA-seq数据解释的组成部分。为了适应对嵌套细胞簇的层次结构的询问,TooManyCells它配备了BirchBeer一个完全可定制的新颖软件,专门为清晰,可解释地显示细胞簇的层次结构而开发(图2a)。BirchBeer是专为显示整个树而设计的,它具有其他一些功能,可帮助用户查找相关种群并促进数据探索。BirchBeer可以根据该子树中的单元格数量缩放每个分支,并使用通过该分支连接的单元格标签颜色的加权平均混合为每个分支点着色(图2a)。混色可以在多个范围内增强对重叠或不同种群的识别(图2b)。叶节点上的单元组可以使用多种可视化格式。叶节点可以显示一个饼状图,显示其细胞组成的分布。为了以单细胞分辨率描述信息,可以在叶节点处对每个单个细胞进行渲染和颜色编码,以可视化其他信息,例如细胞的确切数量,每个细胞上给定基因的表达水平以及其他信息。叶簇(图2b)。为了帮助解释和分析复杂的细胞混合物,birch-tree提供了几种修剪聚类树的方法(图2c-k)。这些方法包括距根的步骤数,最小叶节点大小以及基于分位数的修剪解决方案(图2c)。重要的是,TooManyCells可以在叶节点群集和群集层次结构中的所有嵌套群集之间进行比较。为了帮助选择给定的细胞分区,请BirchBeer提供每个内部或叶节点群集标识号的覆盖图,可用作重叠TooManyCells表达式分析模块的输入,以比较分析不同规模的群集或进行其他下游分析(图2d) 。由于分支缩放宽度在美学上可能是主观的,因此可以对其进行调整(图2e与图2c)或完全禁用(图2f)。由于候选分割的模块性证明了该分裂相对于随机分割模型的重要性,因此BirchBeer可以将模块显示为圆周黑暗程度不同的黑圈,以帮助识别彼此非常不同的种群(图2g)。BirchBeer可以可视化内部和叶节点的多样性(图2h)。除了在树上覆盖连续变量之外,还BirchBeer可以覆盖离散变量。例如,BirchBeer可以覆盖每个细胞和簇的UMI计数(图2i),以及一个或多个基因的表达状态以适应可视细胞分级分析(图2j和2k)。
三、TooManyCells 识别纯细胞簇

簇纯度的比较分析。(a)所分析的种群由x轴(胸腺,脾脏,骨髓,肢体肌肉,舌头,心脏,肺,乳腺,膀胱,膀胱)左侧的x轴上的部位(即器官和组织)的数量表示,肾脏,肝脏}。比较了集群多样性的分布。从TooManyCells分层群集树中考虑了全部,64或32个群集(从根开始的所有步骤,从根开始的6个步骤或从根开始的5个步骤)群集。所有算法均使用默认过滤和参数。较高的多样性表示较低的准确性。(b)逆加权分集定义为逆最小-最大归一化分集和簇大小与单元总数之比的乘积。值越大表示簇越纯净。(c)与(a)相同,不同之处TooManyCells标准化程序:TooManyCells默认标准化(默认标度)项频率-逆文档频率(tf-idf),上四分位数标准化(Upper Quartile Scaled),总和基因中位数计数标准化(Cell和Gene Scaled),然后进行tf-idf标准化。(d)与(b)相同,不同的TooManyCells归一化程序相同。
四、TooManyCells 准确描绘出细胞混合物中的稀有种群
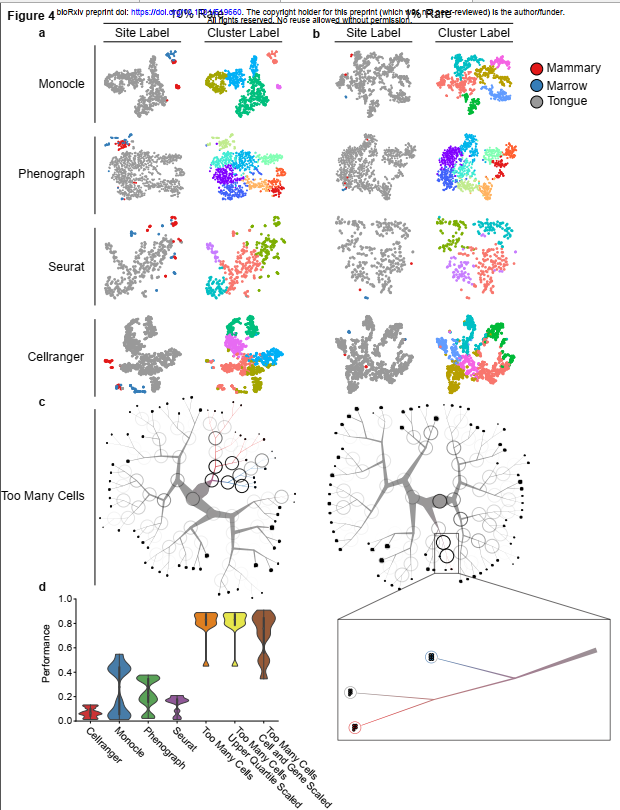
从两个稀有种群与一个普通细胞种群混合中检测细胞,以此作为通用聚类算法(a和b)的基准。从左到右的列:用实际类型(即器官)标记的细胞,以及用scRNA-seq聚类算法分配的簇标记的细胞。从上到下的行:Monocle,Phenograph,Seurat和Cellranger t-SNE投影。提出了对a)900个普通(舌)细胞和100个稀有(50个乳腺和50个骨髓)细胞以及b)990个普通和10个稀有细胞(5个乳腺和5个骨髓)的分析。(c)结果TooManyCells聚类和可视化。左:900(普通)(普通)和100(罕见)。右:990个普通细胞和10个稀有细胞。(d)量化稀有人口基准。对于每次运行,性能代表真正的稀有对(乳腺与乳腺以及骨髓和骨髓在同一簇中)/总稀有对(真正的稀有对和具有骨髓的乳腺)。
五、小鼠脾脏中成浆细胞亚群的鉴定
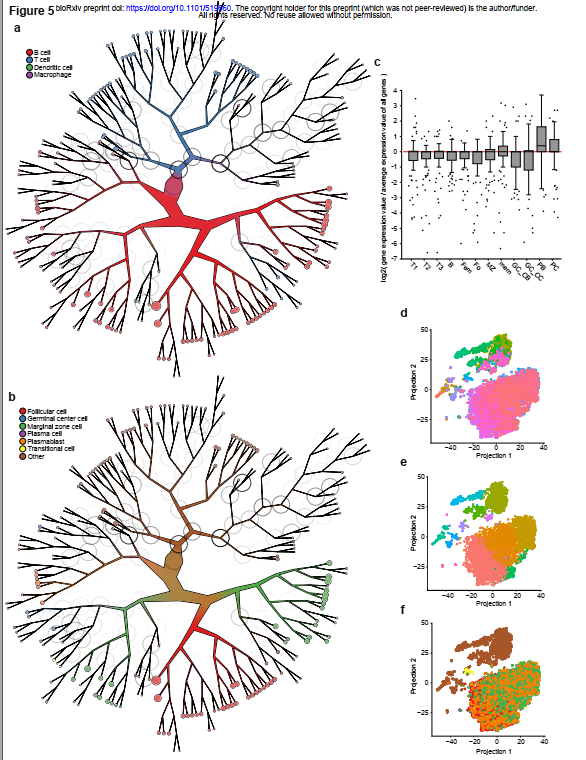
(a) TooManyCells用先前识别的细胞类型标记的小鼠脾脏数据集的聚类树(b)来自(a)的具有新识别的b细胞亚型的树(c)与所有其他细胞相比,来自(b)的成浆细胞节点的前100个差异表达基因的MyGeneSet基因表达(d) (a)中的细胞使用秀兰的加工和t-SNE投影,用TooManyCells对树叶进行聚类,为每片树叶分配不同的颜色类似的颜色表示树中的附近位置,例如,粉色和紫色在树中的位置比粉色和绿色更近(e) (d)中t-SNE投影的坐标,其坐标为(b)中子集的人口成浆细胞是橙色的,分布在整个b细胞群中(f)来自(d)中t-SNE投影的坐标,该投影由修生成的簇标签着色。
六、SOX2OT对体内BCSC生长和致瘤性的影响
SOX2OT对体内BCSC生长和致瘤性的影响。显示了从小鼠收集的肿瘤。b测量并分析了sh-SOX2OT和sh-NC治疗组的肿瘤重量。c测量并分析了sh-SOX2OT和sh-NC治疗组的肿瘤体积曲线。d测量和分析了sh-SOX2OT和sh-NC治疗组的无肿瘤比例。敲低SOX2OT在体内抑制膀胱癌细胞的生长。e和f敲低SOX2OT在体内的BCC中增加了miR-200c的表达,降低了SOX2和SOX2靶基因的表达,并抑制了EMT。G:击倒SOX2OT降低了SOX2的表达,并且SOX2OT和SOX2在BCC体内共定位。h和i击倒SOX2OT会降低体内BCC中SOX2和ki67的表达。j细胞荧光示踪系统的示意图。敲除SOX2OT的k和l显着降低了异种移植物中sh-SOX2OT细胞的比例。数据显示为平均值±SD。* p <0.05;** p <0.01
七、敲减SOX2OT抑制膀胱癌转移
SOX2OT对膀胱癌肺转移的影响以及SOX2OT致癌作用的示意图。a和b sh-SOX2OT组的荧光素酶信号显着低于sh-NC组的荧光素酶信号。c两个治疗组的小鼠体重之间无显着差异。d和e与sh-NC组相比,sh-SOX2OT组的肺转移数目和大小明显减少。f和h降低SOX2OT的表达会降低肺转移中SOX2的表达。G在sh-SOX2OT组中,E-cadherin的表达明显高于在sh-NC组中。敲除SOX2OT可降低肺转移中N-钙黏着蛋白的表达。i SOX2OT在膀胱癌中的致癌作用示意图