






活久见 脂肪能保护小鼠免受中风损害
移植的间充质干细胞(MSCs)在临床前中风模型中通过分泌细胞外囊泡(EVs)可以产生神经保护作用。但是,尚未发现EV装载的神经保护作用物质。为了研究此类物质及其潜在机制,将原代神经元暴露于氧-葡萄糖剥夺(OGD)中,并与脂肪来源的MSC(ADMSC)或分泌了ADMSC的EV共培养。在这种情况下,ADMSC和分泌出ADMSC的EV均可显着减少神经元死亡。进一步发现ADMSC通过EV转移miR-25减少中风小鼠的自噬达到神经保护作用。本文于2020年10月发表在《J Extracell Vesicles》IF:14.976杂志上。
技术路线如下:

主要内容如下:
1、纯化,分离和表征ADMSC-EV
首先使用两种方法UC和PEG从ADMSC中分离出EV,然后使用WB,透射电镜TEM和NTA分析鉴定分离的囊泡颗粒的特征,如图1a-c所示,获得的囊泡颗粒表面表达CD9,CD63,Alix,TSG101,但是不表达albumin,Histone,TOMM20,且UC和PEG两种方法获得EV没有差别,NTA显示大小都在100 nm左右,并具有典型的EV形态,但大小不同。
PEG程序可能无法区分EV与非EV纳米粒子和蛋白质,所以作者接下来鉴定获得的纳米颗粒是来源于UC还是PEG样品。首先使用自形成的碘克沙醇(OptiPrep)对通过UC或PEG分离的EV梯度亚分离(图1d)。分离后对各个亚组分进行WB鉴定,如图1e,UC组和PEG组的EV样品大多以分数7的形式存在。NTA显示,无论选择何种富集方法,绝大多数EV都是分数7。此外,组分7的EV的典型大小是30-150nm(图1g)。因此,因此,碘化醇梯度中可以分离出具有不同浮力密度和大小的EVs亚型,无论采用何种分离方法,小型EVs (sEVs,直径50至200 nm)在第七馏分中富集程度最高。
在随后的实验中,收集了组分7作为纯化的sEVs (iodixanol),用于进一步的实验。尺寸排阻色谱(SEC)被认为是从不同基质中分离纯化EV的最佳方法之一。然后将UC或PEG分离的EV应用于SEC柱过滤后收集。UCSEC和PEG-SEC的特征是基于大小分布和EV富集蛋白的存在。WB分析显示UC-SEC和PEG-SEC都富集了EV标记(如CD9、CD63和Alix),过滤后的和未处理的没有差异(图1h)。图1i给出了基于NTA的具有代表性的UC-SEC和PEG-SEC的尺寸分布曲线,该曲线反映了UC-SEC和PEG-SEC相似的尺寸分布规律,峰值大约在100 nm处。
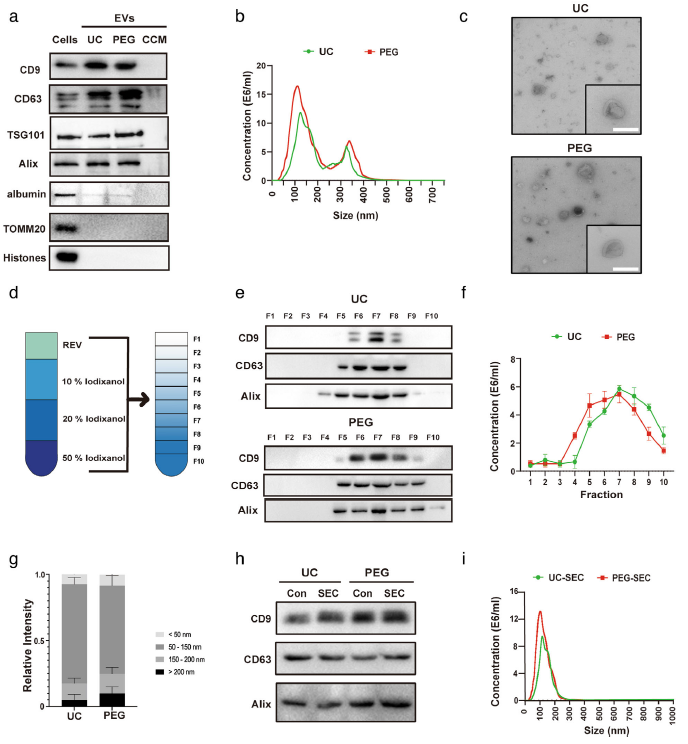
图1 ADMSC-EV的表征和纯化
2、ADMSC-EVs通过调节自噬来保护神经细胞免受氧葡萄糖剥夺(OGD)损伤
随后,作者分析了ADMSC-EVs诱导的神经保护在中风模型中潜在机制。体外模型时将原代神经元暴露于OGD中24小时后再氧化,根据缺血再灌注时间的不同,表现出明显程度的细胞损伤(图2a)。尽管最近的研究表明自噬参与了缺氧或缺血诱导的细胞损伤,但已发表的数据似乎在自噬是有益还是有害这一问题上存在矛盾。因此,作者分析了模型中的自噬水平。其中LC3-II水平与细胞损伤程度显著相关,表现为缺血再灌注10小时的神经元中LC3-II含量明显高于缺血再灌注4小时或8小时的神经元(图2b-c)。之后,所有的实验均采用10小时OGD暴露。与对照组相比,用ADMSCs或UC或PEG分离的EV处理暴露于OGD的原代神经元,细胞损伤显著减少(图2d)。但就存活率而言,EV处理的并不比ADMSCs处理的差。
为了探究ADMSCs来源的EV增强原代神经元细胞对OGD的抗性是否通过调节自噬活性,评估了自噬蛋白的表达。当初代神经元暴露于OGD和与ADMSCs共培养或用ADMSCs来源的EV处理时中风引起的LC3-II蛋白丰度增加的情况被反转了(图2e-f)。表明ADMSC-EVs可能通过调节自噬来保护神经细胞免受OGD损伤。
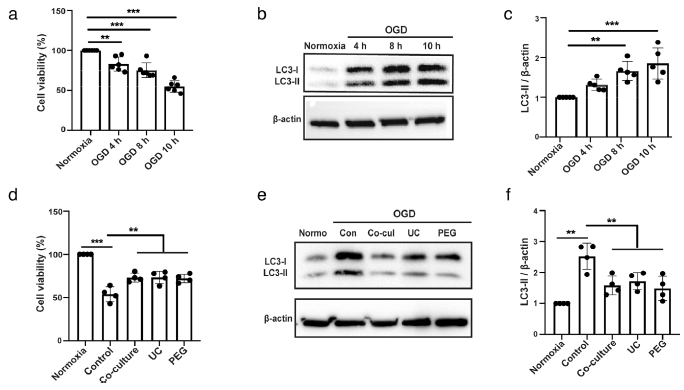
图2ADMSC-EVs通过调节自噬来保护神经细胞免受氧葡萄糖剥夺(OGD)损伤
3、ADMSC-EVs通过p53和BNIP3信号传导抑制自噬通量并增加细胞活力
LC3-II作为自噬的典型标志物,但事实上,LC3-II水平的升高可能是自噬体形成增强的结果,也可能是自噬体与溶酶体融合不足的结果。为了区分自噬过程的这两种状态,通过bafilomycin A1 (BafA1)处理原代神经元来评估自噬的效果,BafA1是一种有效的V - ATP酶抑制剂,可阻碍自噬体-溶酶体融合。如果自噬体的分解被阻断,LC3-II的绝对值积累(处理过的BafA1减去未处理的BafA1)表明在特定时间内产生了多少新的自噬体。与对照组相比,经ADMSCs或EV培养的原代神经元中,BafA1诱导的LC3-II积累量显著降低(图3a-b)。 采用串联荧光RFP-GFP-LC3B报告系统监测原代神经元的自噬通量。用ADMSC-EVs孵育暴露于OGD的原代神经元时,自噬体和自溶溶酶体的数量都减少了(图3c-d)。
为了进一步揭示ADMSC-EVs与自噬的关系,在暴露于OGD的原代神经元中使用自噬激活剂雷帕霉素和抑制剂3-MA(图3e)。不同浓度雷帕霉素与ADMSC-EVs联合处理神经元,可逆转先前EV诱导的神经保护作用,提示ADMSC-EVs与雷帕霉素的作用方式明显相反。相反,使用3-MA以浓度依赖的方式显著增强了细胞的活力(图3e)。表明ADMSC-EVs通过抑制自噬通量并增加细胞活力。
众所周知,自噬调节的4个信号通路包括p53和BNIP3。有趣的是,p53和BNIP3蛋白的表达水平在OGD暴露的原代神经元细胞中都显著升高了(图3f-g)。但是,当与ADMSC或ADMSC-EVs共培养显著降低了OGD诱导的p53和BNIP3表达上调,表明ADMSC-EVs可能通过p53和BNIP3信号调节自噬。
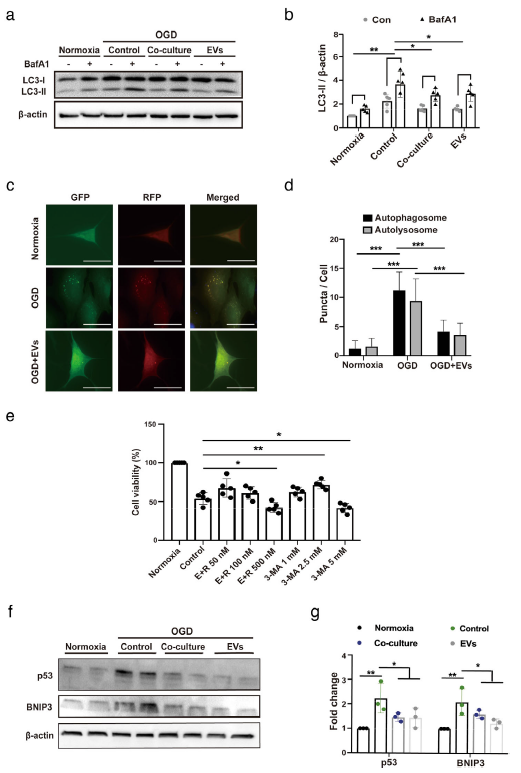
图3 ADMSC-EVs通过p53和BNIP3信号传导抑制自噬通量并增加细胞活力
4、ADMSC-EVs对自噬通量的调节依赖于外泌体
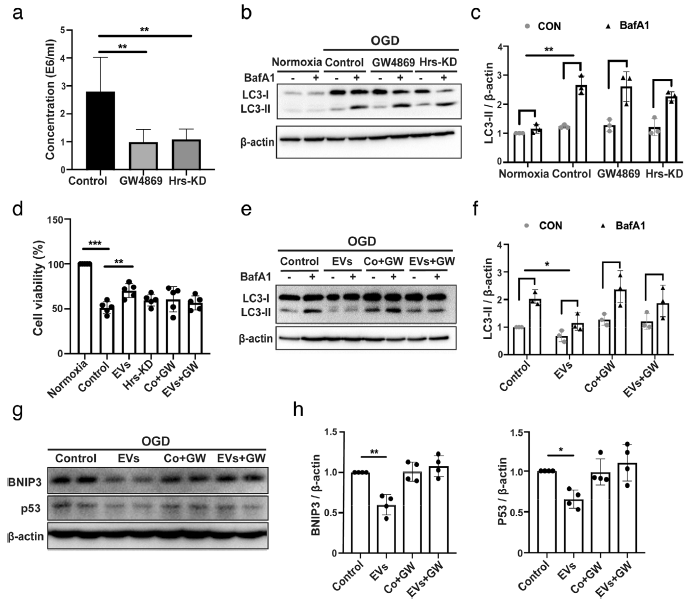
为了确定外泌体在暴露于OGD的原代神经元的自噬调节和神经保护中的作用,用中性鞘磷脂酶靶向抑制剂GW4869抑制神经酰胺合成,并通过敲除ESCRT-0复合物的一个成员Hrs(Hrs-KD)抑制ESCRT介导的外泌体生物发生。如预期地,Hrs-KD和GW4869预处理导致外泌体分泌下降(图4a)。此外,当外泌体分泌抑制剂GW4869或Hrs-KD预处理ADMSC时,ADMSC-EVs (EVs)对自噬调节(图4b-c)和神经保护(图4d)的作用被明显阻断。共培养模型中应用GW4869,当外泌体分泌抑制剂GW4869预处理ADMSCs时,ADMSCs (EVs)对神经保护(图4d)和自噬调节(图4e-f)的作用被明显阻断。重要的是,与ADMSC-EVs共同孵育可降低暴露于OGD的神经元中p53和BNIP3的丰度,而与GW4869预处理的ADMSC获得的EVs共同孵育的神经元不能抑制p53和BNIP3蛋白水平(图4g-h)。因此,形成ADMSC-EV亚群的外泌体似乎在介导暴露于OGD的原代神经元ADMSC的抗自噬活性中起关键作用。
为了鉴定EV介导自噬的实际分子,挖掘了与p53/PTEN作用的miRNA。根据文献可知,有10个miRNA被公认为靶向p53,作者选择了其中了6个用于qRT-PCR,结果发现miR-25-3p在EV中显著高于其它分子(图5a)。为了表明在本实验研究的条件下miR-25的物理化学状态,用RNase A, Triton X-100清洁剂(破坏囊泡的脂质双层),两者都处理EV,或者单独使用溶剂(对照),通过RT-qPCR检测miR-25的水平。结果表明与溶剂处理的信号相比,当用RNase A处理EVs,而不是Triton X-100处理EVs时, miR-25的PCR信号保持在误差范围内。这表明该柱捕获的RNA位于小泡内,因此脂质双分子层保护RNA不被RNase A消化(图5b)。而RNase A和Triton X-100同时使用时miR-25的表达极低,这证实最初对消化的抗性是由于囊泡的隔离,当Triton X-100消化掉保护作用的囊泡膜后,RNase A导致RNA几乎全部被消化(图5b),这证明miR-25在ADMSC-EV内部。然而,当神经元细胞暴露于OGD时,初级神经元中miR-25-3p的细胞内浓度显着降低(图5c)。 相反,用ADMSC-EV治疗原代神经元会显着增加miR-25-3p的浓度(图5c)。
为了证实miR-25-3p的作用,从用anti-miR-25-3p 寡核苷酸预处理的ADMSC(ADMSC-EVanti-miR-25)中分离EV,同时用scramble作对照(ADMSC-EVNC),结果发现,暴露在OGD中后,与PBS预处理对照相比,ADMSC-EVNC共培养的原代神经元的细胞活力显著更高,然而,这些作用在用ADMSC-EVanti-miR-25处理时显著消失(图5d)。与PBS处理的细胞相比,在用ADMSC-EVsNC培养的原代神经元中,在OGD条件下,BafA1诱导的LC3-II积累的量也显着降低(图5e-f),而在用ADMSC-EVanti-miR-25孵育神经元时,这些结果再次反转(图5e-f)。同样,使用萤光素酶系统发现,与ADMSC-EVsNC孵育的神经元相比,ADMSC-EVsanti-miR-25(EVsanti-miR-25)孵育的神经元中自噬体和自溶体的数量显着增加(图5g-h)。总之,这些结果表明ADMSC来源的EV通过miR-25-3p调节自噬并诱导神经保护。

图5 ADMSC通过miR-25-3p调节自噬并诱导神经保护
6、ADMSC-EVs诱导中风的小鼠中持续的神经保护并促进神经恢复
根据上述体外原代神经元数据,进一步探究ADMSC-EV改善中风后神经功能恢复是否通过调节自噬活性。首先检查了缺血条件下EV的生物分布模式。与前面的研究结果一致,ADMSC - EV确实可以到达中枢神经系统,至少在缺血性中风的情况下是这样(图6a)。在再灌注开始时或再灌注后12 h内,全身性注射稀释于200μLPBS中的2×106 ADMSCs释放的EV。与PBS对照相比,在再灌注开始时立即接受ADMSC-EV的小鼠表现出明显更小的梗塞体积(图6b-c)。同样,在12小时内用EV治疗中风小鼠时,梗死面积也明显减少。与对照组相比,两个EV组之间无显着差异(图6b-c)。伴随着这种急性脑损伤的减少,行为测试分析显示,在任一时间点,用EV治疗的小鼠的测试得分更高(图6d-e)。 值得注意的是,在转弯和紧绳测试中,这种更好的测试性能是持久且稳定的,直到14天的观察期结束。随后分析了中风后14天缺血性纹状体的神经元存活情况。与减少神经系统损伤相一致,在两个注射时间点均用ADMSC-EV治疗的小鼠中发现神经元密度增加(图6f-g)。结论是,在实验性卒中模型中,ADMSC衍生的EV在组织学和功能水平上均减少了中风后脑损伤。
7、MCAO后ADMSC-EV给药通过p53-BNIP3信号传导降低自噬通量
为了探究自噬抑制对神经保护的作用,使用3-MA抑制自制在大脑中动脉闭塞(MCAO)后,评估神经元存活和神经恢复。结果发现无论是MCAO后抑制自噬显著增加的细胞存活,但是在中风后12小时3-MA给药处理能更好的促进神经恢复,而在再灌注发生的0小时处理只能部分增强神经恢复(图6d-e)。而在中风后12小时3-MA给药处理神经元密度显著增强,但0小时是估计没有影响(图6f-g)。同样,当在这些动物中同时使用BafA1时,在12 h用3-MA治疗中风小鼠会显着降低手术后24 h的自噬通量,与PBS组相比,这些小鼠的LC3-II水平显着降低(图6h-i)。与此相反,在再灌注开始时0小时立即用3-MA处理对自噬通量没有影响。 这些结果表明,调节中风后过度激活的自噬活性可在MCAO诱导后在小鼠中发挥神经保护作用。
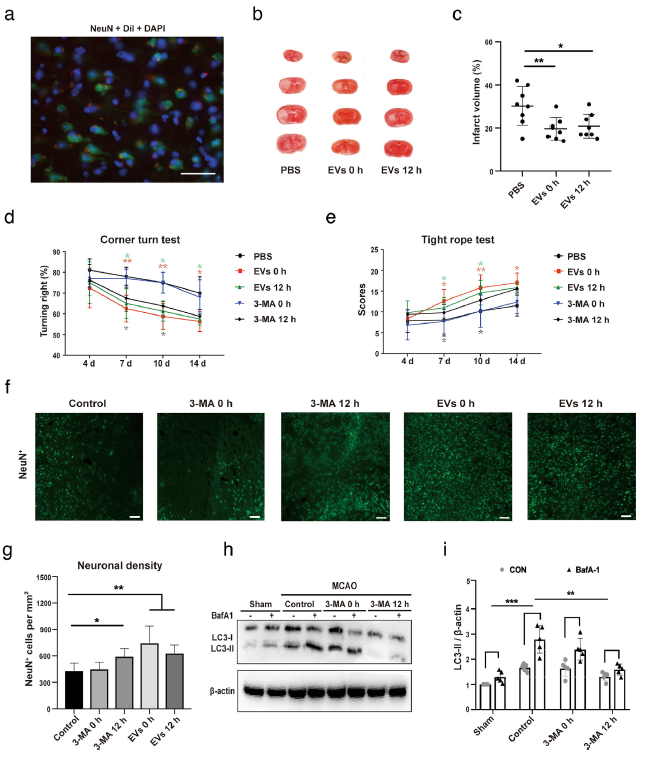
图6 EV引起的自噬调节可减少中风后脑损伤并改善神经功能。
8、ADMSC-EVs中miR-25-3p的丢失减少了中风后EV诱导的自噬和神经保护调节
为了确认与ADMSC-EV给药相关的自噬通量的降低是由miR-25-3p的EV介导的,给小鼠注射了PBS和ADMSC-EV,后者用antimiR25-3p(EVsanti-miR25)预处理的过,或已用对照寡核苷酸(EVsNC)预处理过。注射是在MCAO手术后12小时完成的。 再灌注24小时后,与PBS组相比,ADMSC-EV和ADMSC-EVsNC组中,缺血性纹状体中BafA1诱导的LC3-II积累均显着降低(图7a-b)。但是,ADMSC-EVsanti-miR25的处理未能显示出对自噬通量的类似作用(图7a-b)。
一致地,与对照组和用ADMSC-EVsanti-miR25治疗的小鼠相比,用ADMSC-EVsNC治疗的小鼠通过转弯和紧绳试验评估的神经系统恢复得到增强(图7c-d)。 MCAO 14天后对神经元密度的分析显示,与用ADMSC-EVsanti-miR25治疗的动物相比,用ADMSC-EVsNC治疗的小鼠显示出脑损伤减少。因此,从ADMSC衍生的EV最终可在组织学和功能水平上减少脑损伤。 MCAO后与ADMSC-EV给药相关的抗自噬活性至少部分由miR-25-3p的EV介导。
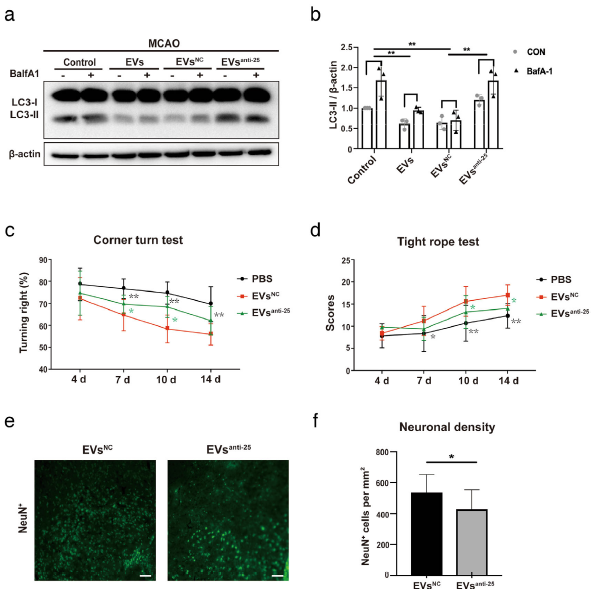
图7 ADMSC-EVs中miR-25-3p的丢失减少了中风后EV诱导的自噬和神经保护调节
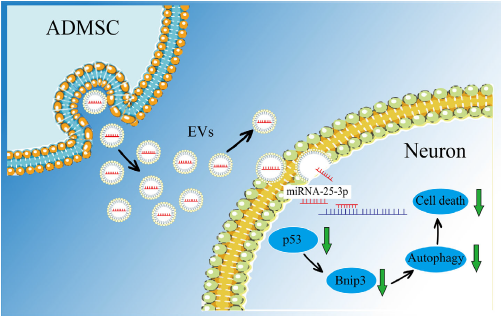
图8源自ADMSC-EV的miR-25-3p在临床前中风模型中的作用示意图。
总之,ADMSC释放富含miR-25-3p的EV,这些EV会被神经元摄取。在神经元中,miR-25-3p诱导p53 mRNA的降解,从而导致p53蛋白水平的下调和随后BNIP3的降低。反过来,对BNIP3的抑制作用会进一步降低自噬水平,从而发挥神经保护作用。
参考文献:
Kuang Yaoyun., Zheng Xuan., Zhang Lin., Ai Xiaoyu., Venkataramani Vivek., Kilic Ertugrul., Hermann Dirk M., Majid Arshad., Bähr Mathias., Doeppner Thorsten R.(2020). Adipose-derived mesenchymal stem cells reduce autophagy in stroke mice by extracellular vesicle transfer of miR-25. J Extracell Vesicles, 10(1), e12024. doi:10.1002/jev2.12024