TYRO3通过抑制天然免疫和铁死亡诱导抗PD-1/PD-L1治疗耐药
导语:免疫检查点阻断疗法已证明对多种癌症类型具有良好的临床效果。程序性细胞死亡蛋白1(PD-1)是免疫检查点,然而,由于耐药性以及用于患者分层的生物标志物不足,仅少数患者从单药治疗中获益,在很大程度上限制了临床效应。这里,小编带大家领略下小分子化合物的在耐药方面的独特魅力。
参考文献:TYRO3 induces anti–PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis (IF=11.864)
技术路线:

结果:
1. TYRO3高表达与抗PD-1/PD-L1治疗患者的预后不良相关
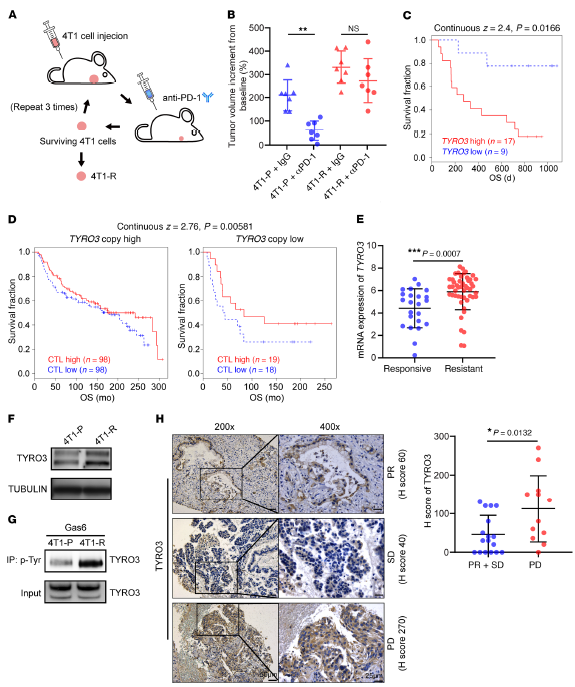
建立肿瘤小鼠体内耐药模型,将4T1乳腺癌细胞接种到BALB/c小鼠的乳腺脂肪垫中,用抗PD-1(抗mPD-1)抗体治疗,发现亲代4T1(4T1-P)肿瘤对抗mPD-1治疗有反应,肿瘤生长减少。相反,耐药的4T1(4T1-R)肿瘤对抗mPD-1无反应。使用市售的RTK抗体阵列系统与4T1-P或4T1-R细胞的裂解物杂交,发现4T1-R细胞中TYRO3、EPHB2、FLT3和TRKA表达或磷酸化水平高于4T1-P细胞,TYRO3的增加最高。在接受抗PD-1抗体治疗的黑色素瘤患者的生存数据中,较高的TYRO3表达水平与较短的总生存期相关,表明TYRO3表达与抗PD-1耐药相关。TYRO3较高的表达与多种癌症类型的较差预后相关。
检测TYRO3基因拷贝数与细胞毒性T细胞抗肿瘤活性的相关性,CD8 + T细胞水平与高表达TYRO3基因的患者的生存期延长无关,但确实介导了低TYRO3基因拷贝数患者的生存期延长,表明TYRO3降低细胞毒性T细胞抗肿瘤作用的观点。
为进一步确定TYRO3和抗PD-1治疗结果之间的相关性,分析接受抗PD-1治疗的黑色素瘤患者RNA-Seq数据中TYRO3的表达,发现耐药肿瘤患者的TYRO3表达水平显著高于肿瘤有反应的患者。Western blot分析也验证了TYRO3,p-TYRO3在4T1-R克隆中的表达增强,说明TYRO3对配体刺激有反应。为进一步确定TYRO3和抗PD-1/PD-L1耐药性在各种癌症类型中是否普遍相关,我们研究了29例继续接受抗PD-1/PD-L1治疗患者的治疗结果。抗PD-1/PD-L1治疗耐药患者的TYRO3表达水平高于对治疗有反应的患者。
综上所述,患者肿瘤组织中TYRO3的高表达和磷酸化与抗PD-1/PD-L1治疗的耐药性相关。
2. TYRO3使肿瘤细胞对抗PD-1治疗耐药
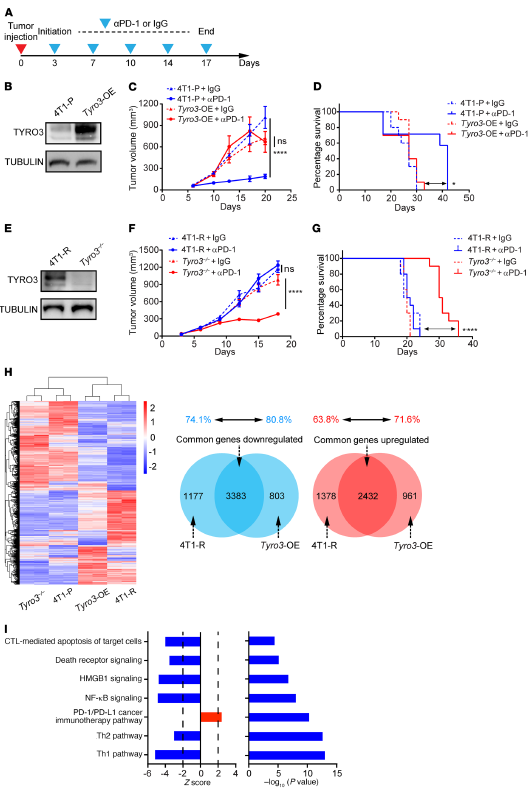
在同系BALB/c小鼠模型中比较了Tyro3过表达(Tyro3-OE)和4T1-P细胞对抗mPD-1治疗的反应。在荷4T1-P肿瘤的小鼠中,抗mPD-1治疗显著减少肿瘤的生长,并延长了生存期。这些结果表明,尽管4T1-P肿瘤对抗mPD-1治疗有反应,Tyro3的过表达促进了这些肿瘤的耐药性。为进一步确定在4T1-R耐药细胞中抗PD-1/PD-L1耐药是否需要TYRO3,敲除4T1-R细胞(TYRO3–/–)中的TYRO3。荷瘤Tyro3敲低细胞的小鼠对抗mPD-1治疗敏感,肿瘤生长显著减少,小鼠存活时间更长。这些结果证明TYRO3在抗PD-1/PD-L1耐药中的重要作用。
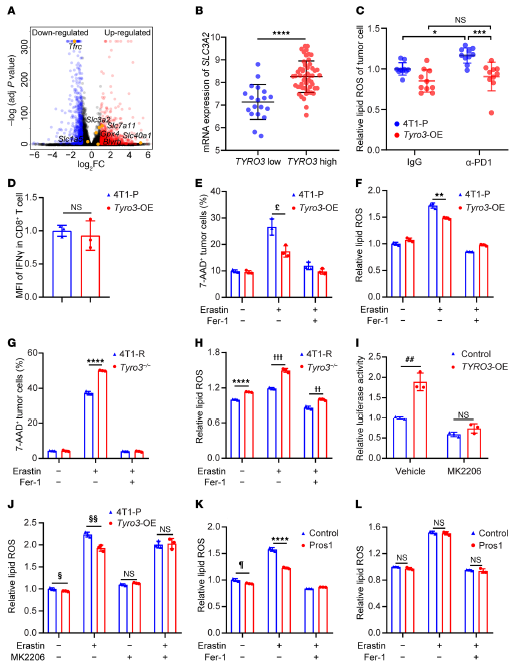
为理解TYRO3在抗PD-1/PD-L1耐药中的机制作用,从4T1-P、TYRO3-OE、4T1-R和TYRO3–/–细胞中提取RNA进行全转录组分析。2种耐药细胞系Tyro3-OE和4T1-R之间的转录组学变化高度相似,变化的重叠百分比较高,表明Tyro3在促进肿瘤细胞对抗PD-1/PD-L1治疗耐药中的作用。来自TCGA数据库的黑色素瘤患者TYRO3基因的通路分析也显示TYRO3表达与PD-1/PD-L1癌症免疫治疗通路呈正相关,与T细胞介导的抗肿瘤反应相关通路(CTL介导的细胞凋亡和死亡受体信号),炎症反应相关通路(Th1活化、Th2活化、NF-κB信号)呈负相关。HMGB1是一种损伤相关分子模式分子,由嗜铁细胞释放,我们发现HMGB1信号与TYRO3表达呈负相关。以上表明,TYRO3在抗PD-1/PD-L1耐药中的作用,并表明TYRO3有利于抗炎TME。
3. TYRO3通过抑制铁死亡和肿瘤TME,诱导抗PD-1/PD-L1耐药性。
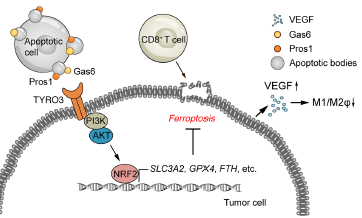
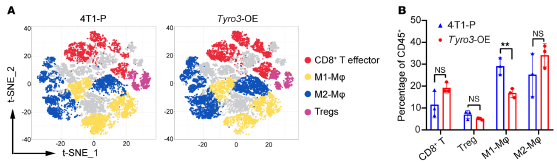
为探究TYRO3在TME中的功能,首先评估了TYRO3-OE 4T1和4T1-P肿瘤之间免疫学特征的整体变化。免疫细胞谱分析发现在CD45-肿瘤细胞中,Tyro3过表达与PD-L1或caspase-3无关,Tyro3-OE肿瘤中M1样巨噬细胞水平降低,M1/M2比值下降,提示肿瘤表达的Tyro3促进M1~M2巨噬细胞极化。用TYRO3-OE BT549细胞或TYRO3-OE和TYRO3–/–4T1肿瘤细胞的条件培养基孵育THP1单核细胞或骨髓源性巨噬细胞,进一步验证TYRO3的体外抗炎功能。RNA-Seq发现VEGF在耐药细胞系4T1-R和Tyro3-OE中表达上调,TYRO3-OE BT549细胞的条件培养基CM显著降低M1标记物的表达,增加M2标记物的表达,加入VEGFR抑制剂完全消除TYRO3对巨噬细胞极化的影响。这些结果表明TYRO3通过上调VEGF降低M1/M2比值,从而促进原瘤TME。
在TYRO3-OE 4T1肿瘤细胞中,阻断铁死亡的基因上调(Slc40a1、Slc7a11、Slc3a2、Gpx4、Fth1和Blvrb),而增强铁死亡的基因下调(Slc5a1、Tfrc)。在接受抗PD-1治疗的黑色素瘤患者中,阻止脂质过氧化和铁死亡的分子SLC3A2与TYRO3显著共表达。抗PD-1治疗后,Tyro3-OE CD45-肿瘤细胞的脂质ROS水平低于4T1-P CD45-肿瘤细胞,而加入抗PD-1显著增加4T1-P肿瘤细胞的脂质ROS,但不增加Tyro3-OE肿瘤细胞的脂质ROS,表明Tyro3抑制抗PD-1诱导的肿瘤细胞铁死亡。在体外用铁死亡诱导剂erastin和铁死亡抑制剂ferrostatin 1(Fer-1)处理4T1-P和Tyro3-OE 4T1细胞。与亲代细胞相比,Tyro3过表达抑制了铁死亡诱导剂诱导的细胞死亡。当Fer-1阻断铁死亡时,Tyro3过表达抑制诱导剂诱导的脂质ROS。Tyro3缺失增加了诱导剂诱导的细胞死亡和脂质过氧化,Fer-1消除了这些作用。这些结果表明TYRO3对于保护肿瘤细胞免受铁死亡至关重要。
NRF2是阻断铁死亡的关键介质,TYRO3过表达的293T细胞中NRF2转录活性增加。TAM激酶下游的PI3K/AKT信号通路可增加NRF2转录活性,TYRO3-OE介导的NRF2转录激活被AKT抑制剂MK2206消除。这些结果表明TYRO3通过AKT/NRF2轴抑制铁死亡。
吞噬细胞对濒死细胞的清除依赖于对凋亡细胞暴露的“吃我信号”的识别。磷脂酰丝氨酸(PS)是一种关键的“吃我”分子,可与桥接分子结合,如TAM激酶配体蛋白S(Pros1)和Gas6。Pros1在与PS结合时同时激活TYRO3。Pros1处理4T1和PY8119肿瘤细胞可抑制erastin诱导的脂质过氧化作用,表明凋亡细胞的Pros1“吃我”信号可以通过TYRO3抑制肿瘤细胞铁死亡。
综上所述,这些发现表明TYRO3抑制肿瘤细胞铁死亡,并通过降低M1/M2比值支持原瘤TME。此外,肿瘤细胞可能利用邻近濒死细胞的Pros1“吃我”信号,通过激活TYRO3,抑制铁死亡,促进肿瘤细胞的存活。
4. 抑制TYRO3可增强铁死亡,使耐药肿瘤对抗mPD-1治疗敏感
TYRO3抑制剂LDC1267处理4T1-R细胞,有效降低了TYRO3磷酸化、,增加肿瘤细胞死亡和脂质过氧化,而在Tyro3-/-细胞中不存在该作用,表明抑制TYRO3可增强肿瘤细胞铁死亡。荷4T1-R肿瘤的小鼠腹腔注射抗mPD-1、LDC1267或其组合,联合给药显著降低了肿瘤生长,并延长小鼠生存期。此外,肾功能和肝功能的生化指标均在其正常范围内,证明联合给药在动物中耐受良好。这些结果表明靶向TYRO3联合抗PD-1有可能克服抗PD-1/PD-L1耐药性,且毒性水平相对较低。
结论:TYRO3抑制肿瘤细胞铁死亡,通过促进M1到M2极化有利于原肿瘤TME,在抗PD-1治疗期间促进肿瘤存活。