






低氧导致肝细胞癌进展的新连接体——CFL1蛋白
肝细胞癌(HCC)是全球第四大致命恶性肿瘤类型。然而,参与HCC进展的确切分子机制尚不清楚。本研究发现缺氧诱导的CFL1通过激活PLD1/AKT通路增加HCC的增殖、迁移、侵袭和EMT。本文于2021年3月发表在《Clinical and Translational Medicine》IF:7.919期刊上。
技术路线:

主要实验结果:
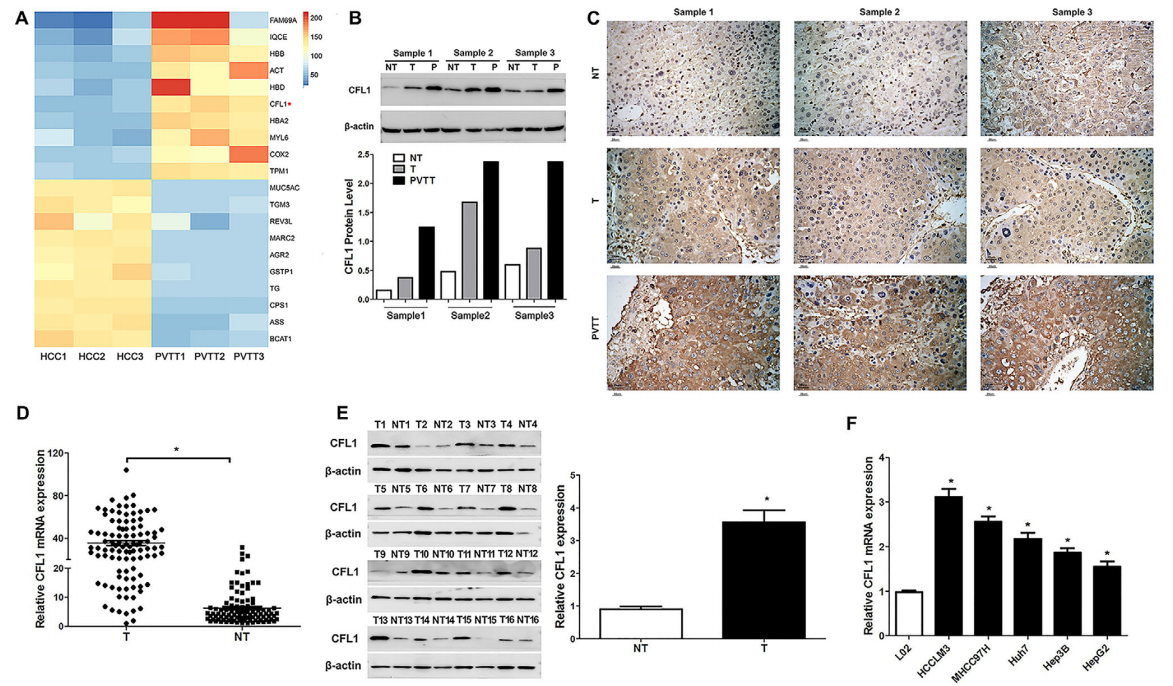
门静脉瘤血栓(PVTT)是HCC预后不良的重要危险因素。取3对HCC和配对的PVTT组织进行iRTAQ检测,挖掘到946个差异表达蛋白。图1A列举了最显著上调的10个蛋白,其中CFL1在HCC中显著高表达。WB和免疫组化表明CFL1在癌旁组织,HCC和PVTT组织中表达逐渐上调(图1B-C)。随后在100对HCC和癌症组织,以及6株细胞系中检测了CFL1的表达,证实CFL1在HCC中高表达。
2、CFL1的高表达与HCC的不良预后相关
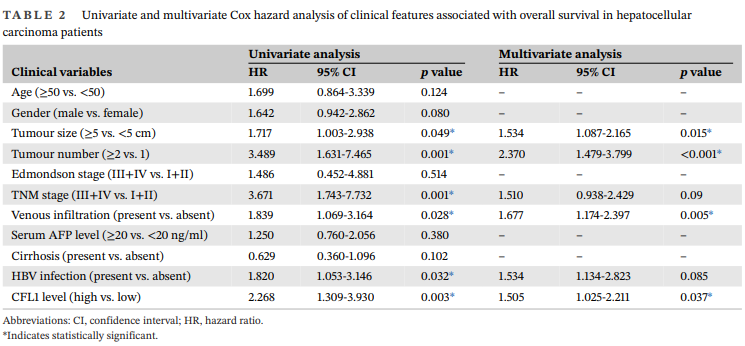
单变量分析显示,肿瘤大小、肿瘤数量、TNM分期、静脉浸润、HBV感染和CFL1水平与HCC患者的总体生存率显著相关(表2)。多变量分析显示,仅肿瘤大小、肿瘤数量、静脉浸润和CFL1水平是HCC总体生存率的独立预后指标(表2)。并且,可以看出CFL1的高表达与HCC的不良预后相关。
3、CFL1促进HCC的增殖,迁移和侵袭
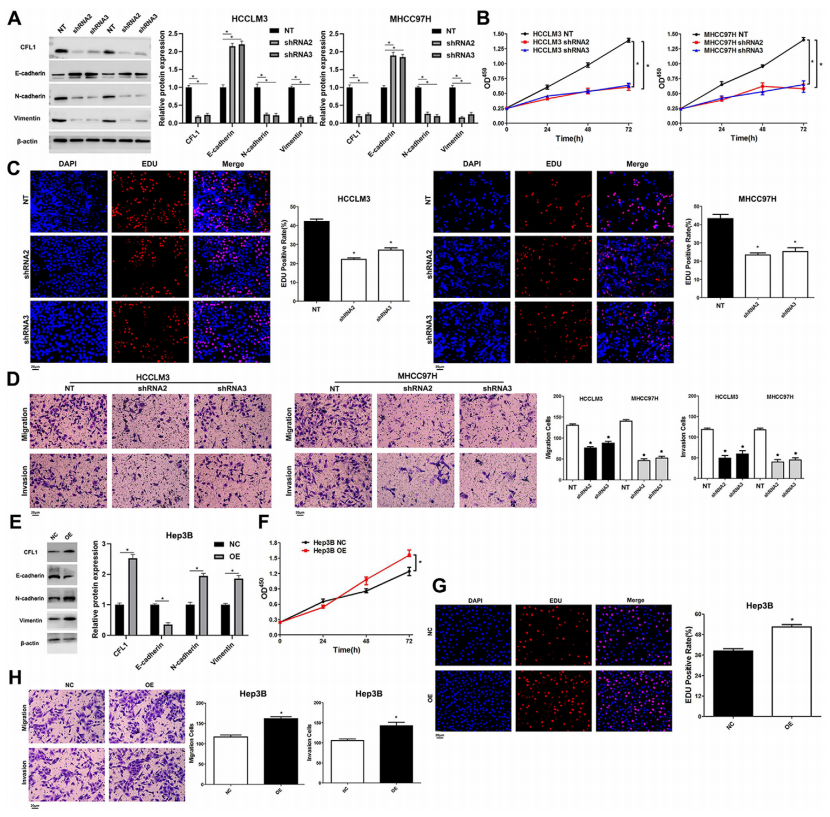
如图2所示,在高表达CFL1的HCCLM3和MHCC97h细胞中,敲除CFL1 (图2A)。随后实验发现敲除CFL1后HCCLM3和MHCC97h细胞的增殖,迁移和侵袭能力被抑制,表明CFL1促进HCC的增殖,迁移和侵袭。
随后,作者在体内进一步验证CLF1的功能。结果如图3所以,敲除CFL1后显著降低了HCC细胞的肿瘤体积和肿瘤,并减少了肺转移结节数量。IHC表明CLF1的敲除也导致EMT相关蛋白的抑制。总之,CLF1敲除抑制小鼠HCC的生长和肺部转移。
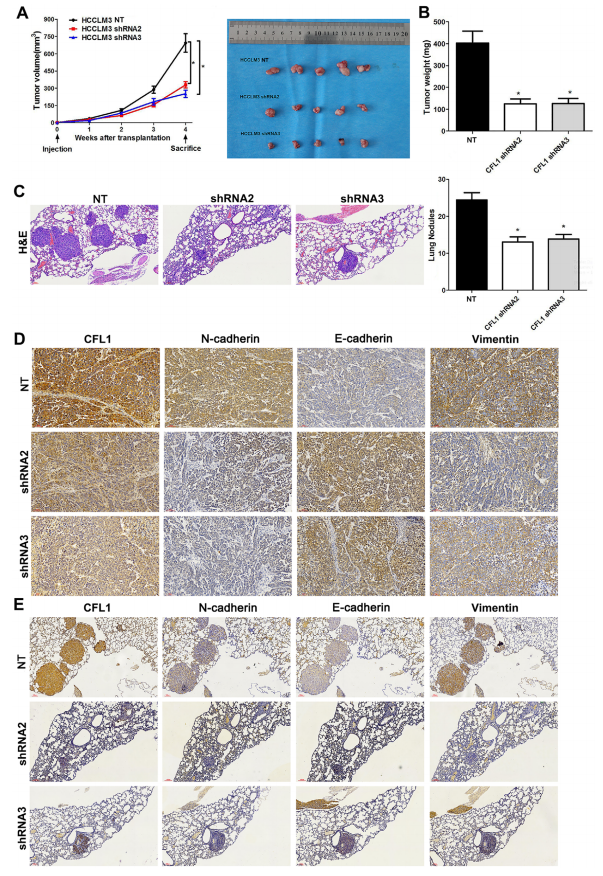
图3 CLF1敲除抑制小鼠HCC的生长和肺部转移
5、CFL1是HIF-1α在HCC细胞中的下游靶基因
为了阐明低氧对HCC中CFL1表达的影响,将HCCLM3和Hep3B细胞在低氧培养箱中培养48 h。结果发现低氧诱导导致CFL1的表达升高,但是当HIF-1α敲除后,低氧诱导并不能提高CFL1的表达,并且使用HIF-1α的抑制剂处理HCCLM3和Hep3B细胞,也能降低因低氧诱导导致CFL1表达升高(图4A-F)。ChIP-PCR检测进一步发现,HIF-1α和HIF-1β与HCC细胞CFL1启动子中的HRE直接结合(图4G)。在低氧条件下,转染HRE荧光素酶质粒或CFL1启动子-荧光素酶质粒的HEK 293T细胞中荧光素酶报告基因活性升高(图4H)。
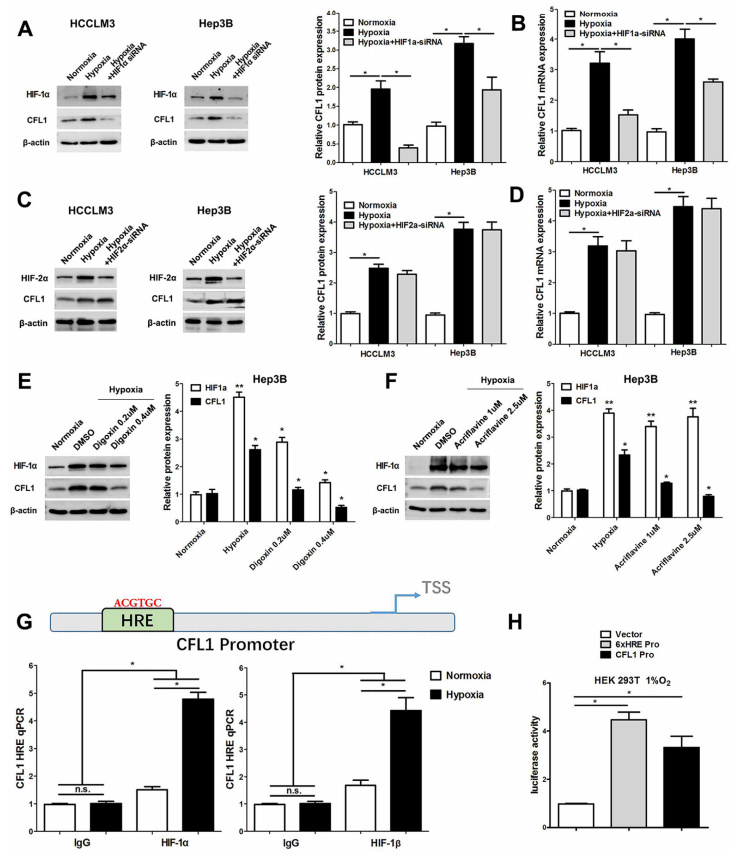
图4 HIF-1α激活HCC细胞中CFL1的转录
接下来,在低氧条件下,在HCCLM3和Hep3B细胞中敲低CFL1的表达(图5A)。结果表明,CFL1敲除显著逆转了缺氧诱导的HCCLM3细胞增殖、细胞迁移和侵袭以及EMT(图5A-D)。综上所述,这些结果表明CFL1表达受HIF-1α的转录调控,介导缺氧诱导的HCC进展。
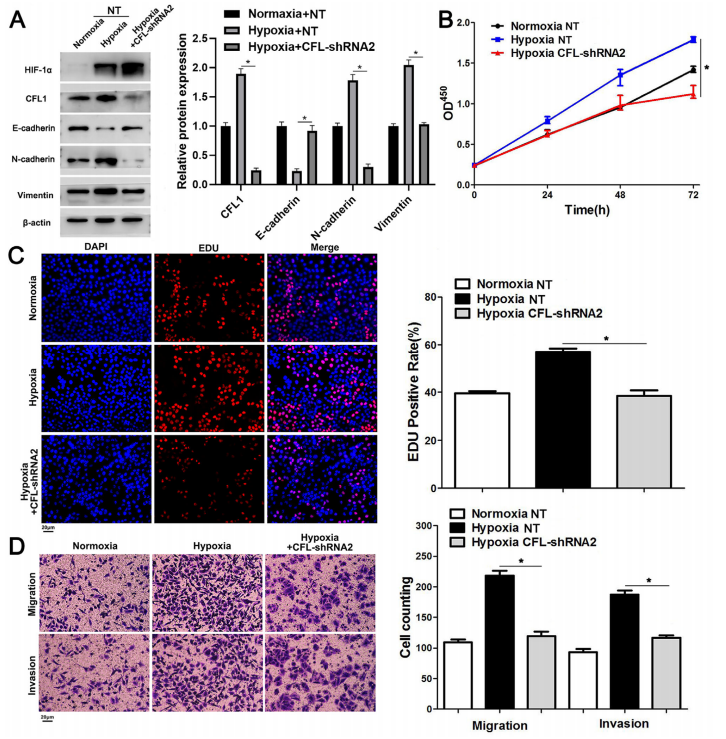
图5 CFL1敲除逆转了HIF诱导的HCC细胞的增殖和侵袭
6、CFL1通过抑制泛素介导的PLD1降解来维持PLD1的表达
为了进一步探索CFL1在HCC中致癌作用的下游机制,利用KEGG数据库筛选了受CFL1影响的通路。其中CFL1的高表达与细胞膜中细胞粘附分子和蛋白的定位密切相关(图6A)。然后,基于蛋白质相互作用数据库预测了CFL1和PLD1之间的潜在相互作用(图6B)。CFL1敲低降低了HCCLM3细胞中PLD1的蛋白水平,而CFL1过表达增强了Hep3B细胞中PLD1蛋白表达(图6C)。co - IP实验表明,CFL1与PLD1相互作用,敲低CFL1可促进肝癌细胞中PLD1的泛素化(图6D-E)。用放线菌酮(CHX,2μg/mL)阻断肝癌细胞蛋白质合成。绘制蛋白降解曲线,表明CFL1敲低时PLD1蛋白降解更快(图6F)。此外,蛋白酶体抑制剂MG132(25 μM)处理可阻断CFL1敲低诱导的HCC细胞中PLD1降解(图6F)。因此,这些结果表明CFL1抑制肝癌细胞中泛素介导的PLD1蛋白水解。
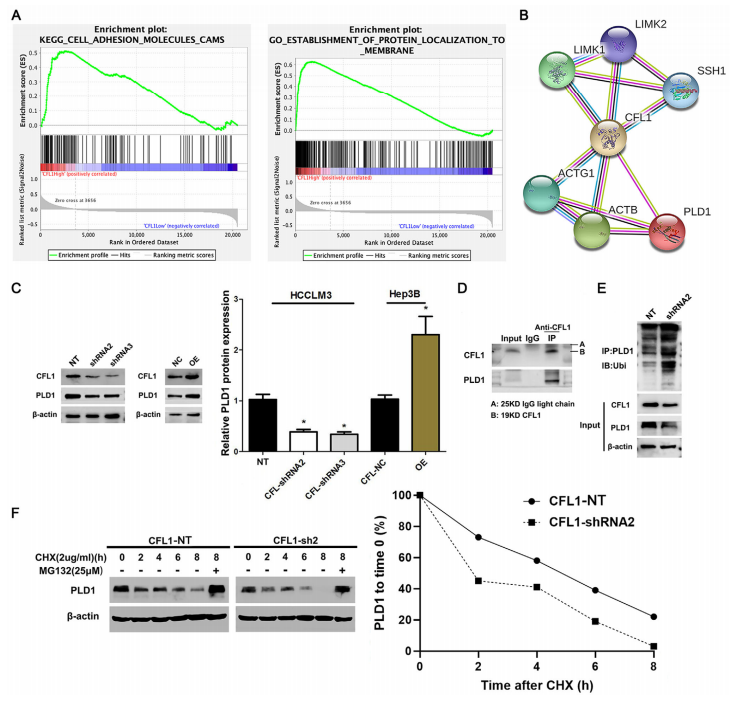
图6 CFL1调节HCC细胞中PLD1的降解
7、CFL1/PLD1轴介导低氧诱导的HCC细胞中AKT途径的激活
之前的一项研究表明,PLD1激活AKT及其下游mTOR通路促进HCC细胞增殖、侵袭。与此一致,作者观察到CFL1敲除降低了p-AKT水平,在HCCLM3细胞中通过PLD1恢复增强了p-AKT水平(图7A,C)。此外,PLD1沉默逆转了CFL1诱导的Hep3B细胞中AKT信号转导通路的活化(图7B,D)。值得注意的是,CFL1或PLD1敲除抑制了肝癌细胞中缺氧诱导的AKT通路激活和EMT过程(图7E,F)。之后,进行拯救实验探讨HIF-1α-CFL1-PLD1轴中PLD1的功能。在HCCLM3细胞中过表达的PLD1可以逆转低氧条件下CFL1-shRNA引起的对AKT磷酸化或EMT过程的抑制(图7G,H)。总的来说,证实CFL1/PLD1轴在缺氧诱导的HCC进展中起重要作用。
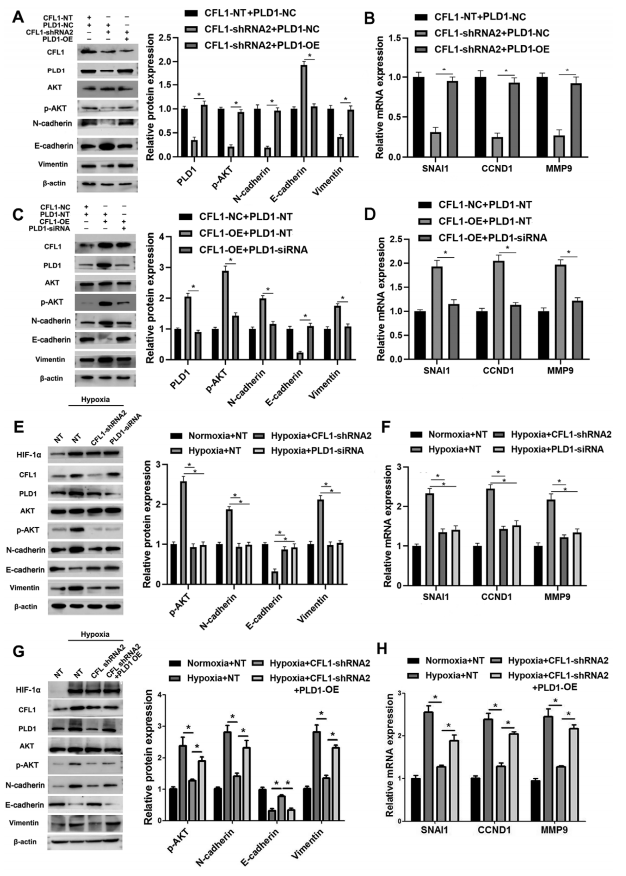
图7 CFL1/PLD1轴介导低氧诱导的HCC细胞中AKT途径的激活
总之,该研究表明HCC中过表达CFL1与较差的临床病理学参数呈正相关。体外和体内实验证实CFL1是HCC肿瘤生长和转移的驱动因素。PDL1/AKT通路介导CFL1的致癌功能。此外,CFL1是一个缺氧反应基因,通过激活PLD1/AKT信号转导参与缺氧诱导的HCC进展(图8)。综上所述,该研究表明CFL1是低氧微环境与HCC进展之间的一种新的连接体。
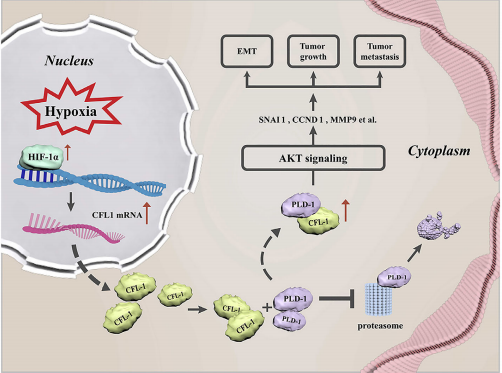
图8本研究结果的示意图
参考文献:
Yao Bowen., Li Yazhao., Chen Tianxiang., Niu Yongshen., Wang Yufeng., Yang Yuanyuan., Wei Xinyu., Liu Qingguang., Tu Kangsheng.(2021). Hypoxia-induced cofilin 1 promotes hepatocellular carcinoma progression by regulating the PLD1/AKT pathway. Clin Transl Med, 11(3), e366. doi:10.1002/ctm2.366