






在乳腺癌中,INPP4B促进pi3k α依赖的晚期核内体形成和Wnt/β-catenin信号转导

磷酸肌醇3-激酶(PI3K)信号通路在癌症中经常过度激活。在胞外刺激下,I类PI3K在质膜内叶上转化为磷脂酰肌醇3,4,5-三磷酸(PI(3,4,5)P3),通过肌醇聚磷酸5-磷酸酶快速转化为磷脂酰肌醇3,4-二磷酸(PI(3,4)P2)。PI(3,4,5)P3和PI(3,4)P2共同增加了致癌PI3K信号的中枢效应蛋白激酶AKT2,3的招募和激活。PI(3,4,5) P3/PI(3,4)P2信号通路激活质膜上的许多其他效应因子,包括蛋白激酶PDK1,该蛋白激酶进而磷酸化AKT和其他AGC激酶,如SGK34。肿瘤抑制因子,磷酸酶和张力蛋白同源物(PTEN),将PI(3,4,5)P3转化为PI(4,5)P2,从而抑制PI3K/AKT信号。II类肌醇多磷酸4-磷酸酶(INPP4B)可使I类PI3K下游的PI(3,4)P2去磷酸化,形成磷脂酰肌醇3-磷酸(PI(3)P),也可抑制AKT信号,并被认为在某些癌症中起抑癌作用。
编码PI3Kα p110α亚基的PIK3CA在多达40%的乳腺癌中发生突变,最常见的是雌激素受体阳性(ER+)肿瘤。突变的PIK3CA在校正ER阳性等有利风险变量时并不是预后的独立标记物,但它是改善晚期ER+乳腺癌内分泌和PI3K联合治疗应答的预测生物标记物。有趣的是,与PTEN等其他PI3K通路改变的肿瘤相比,pikca突变ER+乳腺癌显示出极少的AKT激活和对AKT信号的依赖。
NPP4B抑制AKT信号,并在三阴性(ER /PR /HER2)和基底样乳腺癌中表现出肿瘤抑制活性。此外,INPP4B蛋白在黑色素瘤、卵巢癌和前列腺癌中表达减少。在小鼠模型中,Inpp4b消融与Tp53/Brca1缺失共同增加了乳腺肿瘤外显率,并与Pten杂合子缺失共同促进体内甲状腺肿瘤的发生和转移。值得注意的是,INPP4B通过降解PI(3,4)P2,抑制早期核内体的局部AKT2信号通路和EGFR降解。
最近的研究表明INPP4B可能在与化疗耐药性相关的急性髓系白血病(AML)中起致癌作用,以及在黑素瘤和结肠癌中起致癌作用。而且可能与环境有关。例如,在乳腺癌中,SGK3被扩增,它的激酶活性依赖于致癌的PI3K和INPP4B。最近,INPP4B被确定为与ER+乳腺癌和肿瘤分级相关的顶级基因。
2021年5月,在Nature communications 杂志上发表了文章“INPP4B promotes PI3Kα-dependent late endosome formation and Wnt/β-catenin signaling in breast cancer”。此报道探讨了INPP4B在ER+乳腺癌中的作用,揭示了一组pik3c突变ER+乳腺癌表现出INPP4B表达增加。尽管同时抑制AKT信号,但过表达INPP4B可以促进pik3a突变ER+乳腺癌细胞系的增殖和肿瘤生长。为了研究这一明显的悖论,使用了蛋白质组学、转录组学和先进的细胞成像的综合方法来阐明INPP4B促进pik3a突变ER+乳腺癌发生的分子机制。结果显示INPP4B刺激晚期核内体上pi3k α依赖的信号枢纽,指导Wnt/β-catenin的激活。这些研究揭示了促进细胞增殖和肿瘤生长的两种致癌信号通路之间的串扰机制。
技术路线:

一、在pik3a突变的ER+乳腺癌中,INPP4B的表达增加

INPP4B被确定为人类乳腺癌er阳性标记物.INPP4B在三阴性乳腺癌中表达缺失,然而,它作为其他癌症中潜在的致癌基因的出现,促使我们检测其在ER+乳腺癌中的相对表达和功能。INPP4B蛋白表达缺失与三阴性乳腺癌相关(图1a, b) 增加INPP4B蛋白表达在14 40%的乳腺癌相对于正常组织与ER / PR-positivity和腔内(ER +和/或PR+)乳腺癌亚型(图1 a, b和补充图1 b, c)。在mRNA表达数据不能用于这些cohorts使用组织扫描乳腺癌cDNA阵列I IV (OriGene)检测130例原发人类乳腺癌和16例正常乳腺组织的INPP4B mRNA表达(图1c)。INPP4B表达降低与三阴性乳腺癌相关,而显著增加的INPP4B表达在25%的乳腺癌中与ER/ propositivity和luminal亚型相关(图1c, d和补充图1d, e)。使用METABRIC和TCGA数据集,我们发现只有1%的乳腺癌表现出INPP4B基因改变,如突变、截断、扩增或缺失。INPP4B mRNA表达与PTEN或AKT1突变无关,但与pik3a突变状态正相关(图1e和补充图1f)。在PIK3CA多突变的乳腺癌中,INPP4B的表达也显著升高(补充图1g)。进一步分层研究发现,INPP4B高表达与pik3c突变ER+乳腺癌亚型特异性相关(图1f)。
二、INPP4B增强pik3a突变ER+乳腺癌和乳腺上皮细胞增殖

GFP-INPP4B在MCF-7和T47D ER+乳腺癌细胞中表达,这两种细胞分别有PIK3CAE545K和PIK3CAH1047R高活化突变.癌细胞进行非锚定细胞生长的能力是细胞转化的一个标志。GFP-INPP4B显著增加了软琼脂中MCF-7细胞集落大小(2.1倍)和T47D细胞集落大小(1.8倍),但对集落数量没有影响(图2a-c)。与细胞增殖增加相一致。与载体对照相比,GFP-INPP4B在血清剥夺后增强了MCF-7细胞的增殖(1.7倍)(图2d, e),但不影响在含血清培养基中生长的MCF-7细胞的增殖。过表达GFPINPP4B的MCF-7细胞中,egf刺激的AKTS473和AKTT308磷酸化水平略有降低(图2f-h)。表达MCF-10A腺泡的Myc-INPP4BWT而不是Myc-INPP4BC842A (PI (3,4)P2 4-磷酸酶死亡)的腺泡(补充图2k)显示增殖细胞的百分比明显高于病媒控制的腺泡(1.5倍)(图2i, j)。在第7天,腺泡的大小没有明显变化(图2k)。第14天,Myc-INPP4BWT而不是MycINPP4BC842A表达MCF-10A的细胞形成了更大的腺泡(1.8倍),每个腺泡的细胞数量比载体对照增加了1.4倍(图2l n)。过表达INPP4B促进MCF-10A细胞增殖,但不影响细胞的尖基底极性和细胞凋亡。与此一致的是,过表达Myc-INPP4BWT而不是Myc-INPP4BC842A在血清剥夺后的单层培养中增强了MCF-10A细胞的增殖(1.7倍)(图2o, p)。
总之,这一数据与INPP4B以PI(3,4)P2 -磷酸酶依赖的方式促进pik3a突变体ER+乳腺癌和pik3a野生型乳腺上皮细胞增殖的解释相一致。
三、INPP4B促进晚期核内体上PI(3,4)P2到PI(3)P的转化
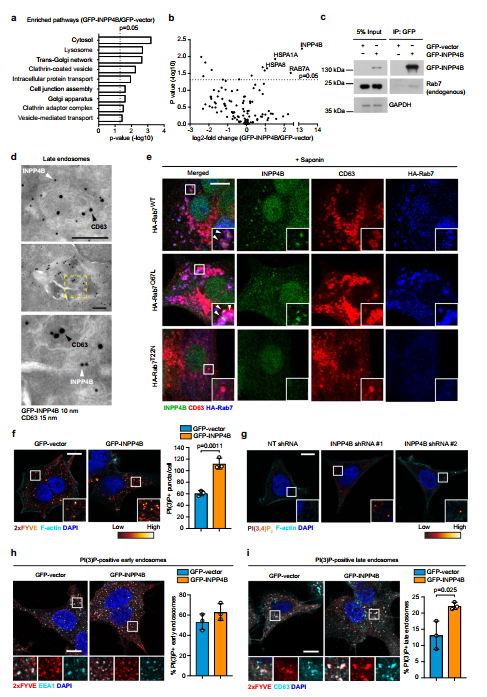
使用了几种基于无偏性蛋白质组学的技术来表征INPP4B在pik3a突变ER+乳腺癌中的下游信号功能。首先,利用液相色谱-串联质谱(LC-MS/MS)进行全细胞蛋白质组学研究,确定GFP-INPP4B与gfp载体表达MCF-7细胞中蛋白水平的差异(2倍)。功能注释分析显示,inpp4b过表达细胞中上调的蛋白与内溶酶体系统密切相关,内溶酶体系统是介导细胞外受体和货物内化和降解的囊泡运输途径(图3a)。免疫沉淀质谱(IP-MS)对受EGF刺激的MCF-7细胞进行GFP-INPP4B蛋白复合物的分析,以激活I类PI3K信号。从该分析中鉴定出的最富集的结合伙伴是RAB7A (Rab7),这是一种定位于晚期核内体的小GTPase(图3b)。GFP-INPP4B与内源性Rab7的共免疫共沉淀证实了INPP4B与Rab7的结合(图3c)。通过免疫电镜进一步分析INPP4B定位于晚期核内体,结果显示GFPINPP4B定位于晚期核内体的限制膜和内膜(图3d),之前的研究已经确定了PI(3,4)P2和PI(3)P。在皂苷处理的MCF-7细胞中,观察到内源性INPP4B与HARab7WT或HA-Rab7Q67L共同定位于cd63阳性的晚期核内体(图3e)。HA-Rab7T22N是一个不定位于晚期核内体的显性阴性突变体(补充图3f),它的表达也阻止了内源性INPP4B的募集(图3e),表明活性的gtp结合的Rab7是INPP4B亚细胞定位到晚期核内体所必需的。 GFP-INPP4B显著提高了细胞内PI(3)P水平(1.8倍)(图3f),PI(3,4)P2也存在于早期和晚期核内体。表达INPP4B shrna的MCF-7细胞显示出更明显的细胞内PI(3,4)P2斑点,表明INPP4B缺失抑制PI(3,4)P2在核内体上的降解,导致其积累(图3g)。GFP-INPP4B没有改变PI(3) p阳性早期核内体的比例,但显著增加了PI(3) p阳性晚期核内体的比例(1.7倍)(图3h, i)。
四、INPP4B促进pi3k α依赖的晚期核内体形成
五、阻断atm依赖的磷酸化会损害DNA损伤后PTEN的亚细胞再分配
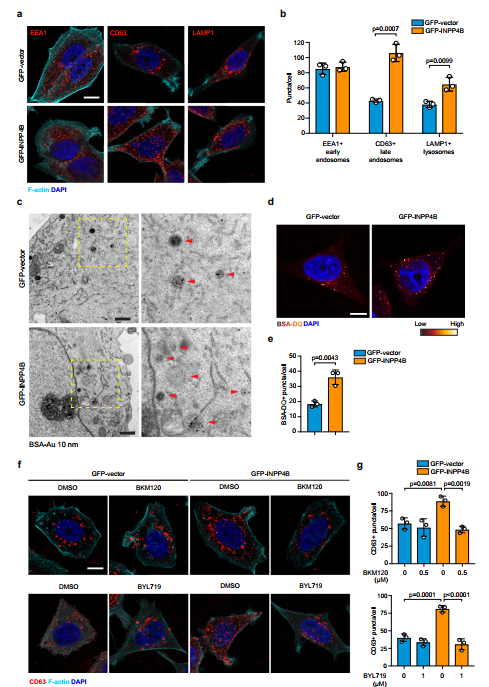
对MCF-7细胞内溶酶体室的检查显示,GFP-INPP4B不影响eea1阳性的早期内溶体,但显著增加了cd63阳性的晚期内溶体(2.5倍)和lamp1阳性的溶酶体(1.7倍)(图4a, b)。
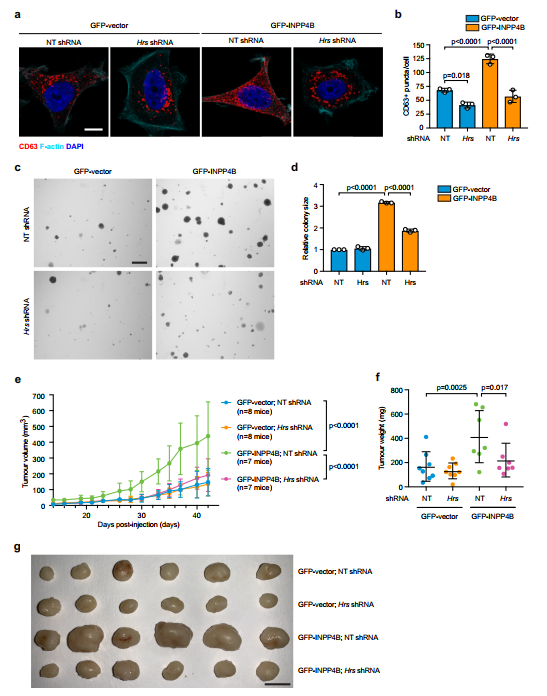
提示INPP4B促进晚期内吞体/溶酶体的形成。使用bsa -gold标记MCF-7细胞内溶酶体室,在电镜下进行超微结构分析。GFP-INPP4B增加了类似晚期核内体的bsa阳性空泡结构的数量(图4c)。利用染料淬灭的BSA (BSA- dq)检测了运往溶酶体的货物运输,BSA- dq通过液相内吞进入内吞体,溶酶体水解酶随后促进去淬和激活荧光信号。GFP-INPP4B显著增加了bsa - dq阳性结构的数量(2倍)(图4d, e),表明INPP4B增强了内吞货物通过晚期内吞体到溶酶体的运输。相比之下,使用泛I类PI3K抑制剂BKM120、PI3Kα特异性抑制剂BYL719(补充图5f)或使用两种不同的siRNAs耗尽PIK3CA抑制GFP-INPP4B细胞中晚期核内体形成的增加(图4f, g和补充图5i, j)。通过在晚期核内体上产生PI(3)P, INPP4B通过Hrs促进晚期核内体的形成。显著的shrna介导的Hrs耗散(补充图6a, b)导致gfp载体细胞中cd63阳性晚期核内体减少,并将GFPINPP4B细胞中晚期核内体形成增加的水平逆转为gfp载体控制水平(图5a, b)。在非锚定细胞生长试验中,耗用Hrs shRNA对gfp载体软琼脂集落的大小没有影响,但减少了GFP-INPP4B细胞集落的大小,这与Hrs依赖的细胞增殖一致(图5c, d)。将MCF-7细胞注射到雌性BALB/c裸小鼠腹股沟第四乳腺脂肪垫中,在体内检测肿瘤生长的hrs依赖性。与GFPvector相比,GFP-INPP4B在体内肿瘤体积和体外肿瘤重量显著增加(3倍)(图5e g)。虽然敲除Hrs没有影响gfp载体肿瘤的大小和重量,但GFP-INPP4B肿瘤的增强生长被减少Hrs抑制(图5e g),表明INPP4B促进pik3a突变ER+乳腺癌细胞的增殖和肿瘤生长以hrsdependence的方式。
六、INPP4B通过增强GSK3β的溶酶体降解促进pi3k依赖的Wnt/β-catenin信号转导
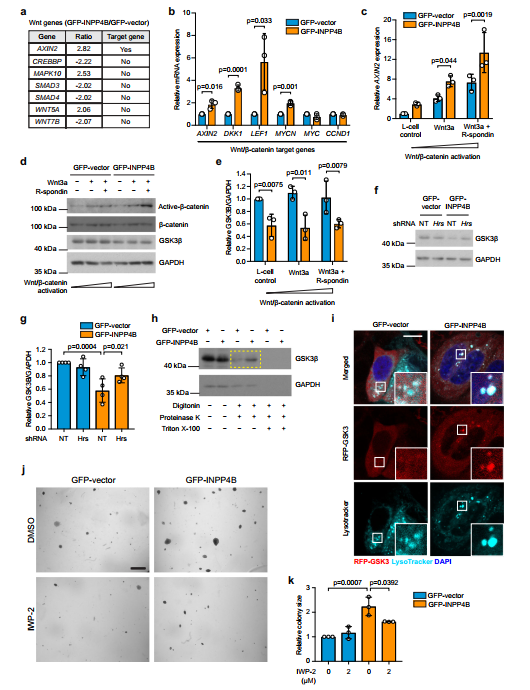
通过nanoString RNA谱分析,我们检测了GFP-INPP4B和gfp载体表达MCF-7细胞中已知的癌症通路,结果显示几种Wnt/β-catenin通路基因的表达发生了改变(图6a)。定量RTPCR证实GFP-INPP4B-MCF-7细胞中Wnt/β-catenin靶基因AXIN2(1.8倍)、LEF1(5.6倍)、DKK1(3.3倍)和MYCN(2倍)的mRNA表达增加,表明Wnt/β-catenin信号通路增加(图6b)。它结合TCF/LEF转录因子,促进Wnt靶基因的转录。Wnt/β-catenin信号通路被Wnt3a和R-spondin处理激活,在这些条件下,与gfp载体对照相比,GFP-INPP4B细胞表现出AXIN2 mRNA表达和非磷酸化-β- catenins33 /S37/T41(活性-β-catenin)水平增加(图6c, d)。此外,GFP-INPP4B的表达在基础和wnt刺激条件下降低了GSK3β蛋白水平,与GSK3β的增强破坏一致(图6d)。通过Hrs shRNA消耗抑制晚期核内体形成后,降低的GSK3β蛋白水平得以恢复(图6f, g)。GFP-INPP4B而不是gfp载体细胞表现出明显的GSK3β蛋白酶保护,这与GSK3β向晚期核内体转运的增强相一致(图6h)。免疫荧光证实了这一点,在GFP-INPP4B中观察到RFP-GSK3与LysoTracker共定位,这是一种积累在晚期核内体/溶酶体中的染料,而不是gfp载体细胞(图6i)。
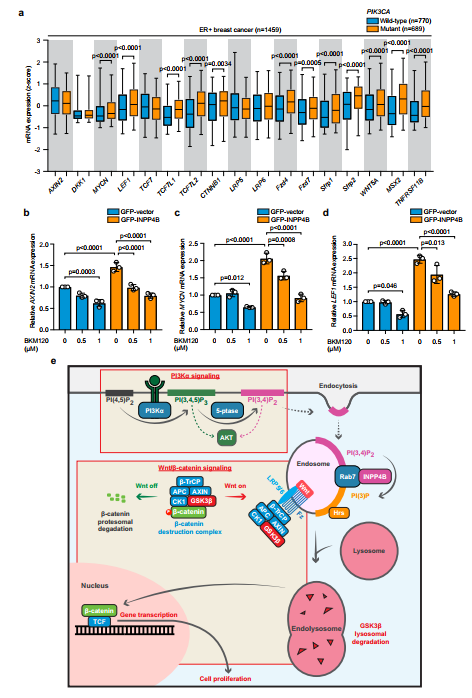
利用METABRIC数据集,我们评估了1459例ER+乳腺癌中17个常见Wnt/β-catenin通路基因的表达,包括几个Wnt靶基因(图7a)。为了确定INPP4B是否促进PI3K和Wnt/β-catenin通路之间的对话,我们用增加剂量的PI3K抑制剂BKM120(0.5和1 M)处理gfp载体和GFPINPP4B MCF-7细胞,并测量Wnt靶基因AXIN2、LEF1和MYCN的mRNA表达(图7b d)。在gfp载体细胞中,低剂量的BKM120对Wnt靶基因表达的影响最小,而高剂量的Wnt基因表达降低。在GFP-INPP4B表达细胞中,低浓度的BKM120部分挽救了Wnt基因表达的增加,在更高浓度时进一步降低。总之,这些发现与INPP4B促进Wnt/β-catenin信号转导,从而通过增强晚期核内体形成促进pik3a突变ER+乳腺癌细胞的增殖的解释相一致(图7e)。