






在HER2阳性乳腺癌中,PTEN和ATM轴控制G1/S细胞周期检查点和肿瘤发生

PTEN包含一个n端磷酸酶结构域,它可以使脂质细胞膜的一种成分——磷脂酰肌醇3,4,5-三磷酸(PI(3,4,5)P3或PIP3)去磷酸化。PTEN通过去磷酸化PIP3的D3位,拮抗磷脂酰肌苷3-激酶(PI3K)通路。PI3K通路调节多种细胞过程,包括细胞代谢、存活、增殖、凋亡、生长和迁移。这些基本的细胞过程,当解除控制时,可以促进或驱动恶性表型。
PTEN功能突变的体细胞丢失在多种人类癌症中被发现,包括乳腺癌、子宫内膜癌、多形性胶质母细胞瘤、皮肤癌和前列腺癌。PTEN缺失是乳腺癌中常见的事件,与加速进展和不良预后密切相关。特别是PTEN的表达已经被提出在人表皮生长因子受体2 (HER2)过表达乳腺癌中发挥重要作用。HER2是表皮生长因子受体家族的一员,具有酪氨酸激酶活性。它的过表达,在大约15 - 20%的乳腺癌病例中观察到,与侵袭性临床行为和不良预后相关。曲妥珠单抗是一种与HER2胞外结构域具有高亲和力的单克隆抗体,是一种有效的治疗HER2阳性乳腺癌患者的方法。PTEN表达检测的总有效率约为70%,而PTEN表达阴性患者的总有效率仅为20%。
PTEN功能的丧失通常与基因组的不稳定性有关。此外,小鼠胚胎成纤维细胞(mef)中PTEN基因的缺失导致了未修复的DNA双链断裂的积累。PTEN缺失被认为通过至少两种分子机制促进了基因组的完整性。在细胞核中,PTEN与着丝粒结合蛋白CENP-C结合,促进着丝粒组装和中期到后期的转变。此外,作为转录因子E2F1的辅助因子,核PTEN似乎调节Rad51的表达,Rad51是DNA修复机制的关键组成部分。然而,后续的研究得出了不一致的结果,表明PTEN在转录水平上对RAD51的调控可能仅限于特定的细胞环境。
PTEN缺陷改变了多个细胞周期检查点,可能留下更少的时间进行DNA损伤修复和/或染色体分离。细胞周期的进展需要几个分子过程的完美执行,以确保一个熟练的,无错误的,细胞分裂。这些事件发生的速度由周期蛋白依赖激酶(CDKs)的活性决定,CDKs磷酸化关键底物以促进DNA合成和有丝分裂进程。CDKs的催化活性受细胞周期检查点的调控,这些检查点监测细胞周期中主要事件的有序执行。检查点代表了故障安全机制,它确保只有在满足的最佳情况下才允许细胞分裂。所有生物体都需要通过细胞分裂周期进行适当的基因组维护,以确保正常生殖、发育和预防包括癌症在内的各种疾病。DNA损伤可由内源性过程引起,如DNA复制过程中偶尔引入的DNA不匹配,拓扑异构酶I和拓扑异构酶II活性失效导致的DNA链断裂,或由正常代谢副产品产生的ROS攻击DNA。外源性来源主要包括诱变化学品、紫外线和电离辐射(IR)。细胞周期检查点能够检测DNA损伤,提示其存在,并激活延缓细胞周期进程的通路,修复DNA损伤,或通过诱导细胞死亡来消除基因不稳定细胞。
哺乳动物细胞DNA损伤反应(DDR)信号的核心是共济失调毛细血管扩张症突变(ATM)和ATM-和rad3相关(ATR)蛋白激酶。ATM和ATR磷酸化并激活另外两个激酶CHK1和CHK2, CHK1和CHK2与ATM和ATR一起,是细胞周期检查点[24]的主调节器。通过调节CDKs的活性,这些分子在细胞周期G1 S期、S内期和G2 M期减缓或阻止细胞周期进程,使DNA损伤得以修复。ATM和ATR通过控制不同因子在DNA损伤位点的表达、活性或招募来促进DNA修复。一般来说,DDR机制能够修复细胞在其生命周期中积累的DNA损伤,但如果认为损伤程度过高,就会诱导细胞凋亡或细胞衰老导致细胞死亡。
2021年4月,加拿大University Ave和中国香港大学团队与癌症研究所表观遗传学和基因组稳定性小组在Cell Death & Differentiation 杂志上发表了文章“The PTEN and ATM axis controls the G1/S cell cycle checkpoint and tumorigenesis in HER2-positive breast cancer”。此报道发现ATM磷酸化PTEN的398位(人类苏氨酸;在小鼠丝氨酸)的激活,为了理解ATM磷酸化PTEN的生物学意义,建立了一个小鼠模型,构建了一个不能被ATM磷酸化的PTEN突变形式,用398位(PTEN- 398a)的丝氨酸取代了丙氨酸。PTEN-398A的表达可加速her2阳性乳腺癌小鼠模型的肿瘤发展和进展。利用分子生物学方法,发现了一种新的机制,通过ATM磷酸化PTEN调节其细胞再分配,并有助于该蛋白的肿瘤抑制功能。
技术路线:

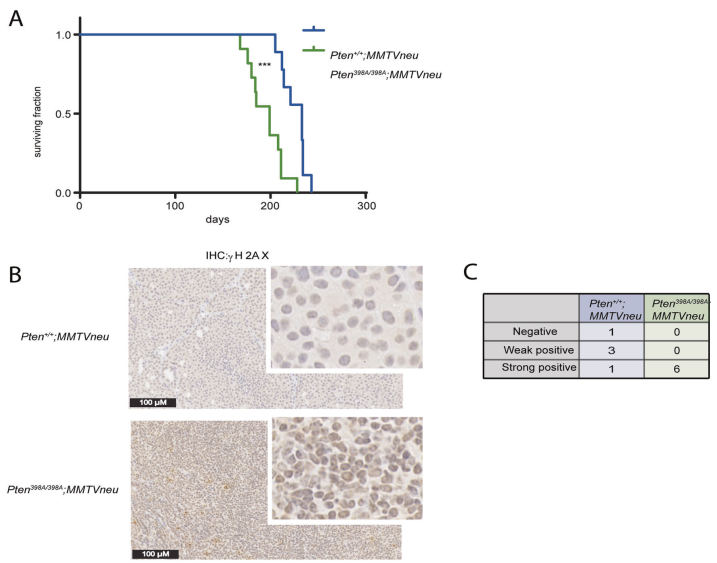
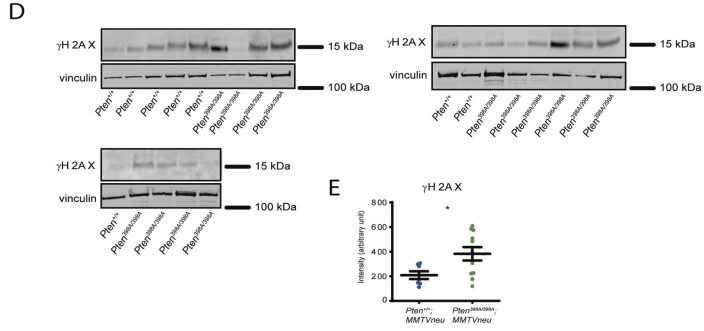
为了研究ATM磷酸化PTEN的体内功能,我们在小鼠中设计了一个敲入等位基因,其中398位丝氨酸取代了丙氨酸(Pten398A)。Pten398A/+和Pten398A/398A小鼠存活,发育明显正常。为了研究atm依赖的PTEN磷酸化在乳腺癌中的可能作用,我们在小鼠乳腺肿瘤病毒(MMTV)启动子/增强子转录控制下表达灭活neu (Erbb2)融合基因的小鼠背景下培养了Pten398A等位基因。在这个成熟的模型中,MMTVneu基因在正常乳腺上皮中低水平表达,导致乳腺肿瘤的发展,据报道中位发生率为205天。Pten+/+;MMTVneu小鼠发生了预期潜伏期的乳腺肿瘤,显示了233天的中位生存期(图1A)。相比之下,Pten398A/398A;MMTVneu小鼠肿瘤发展加快,中位生存期缩短至199天(图1A)。对Pten+/+;MMTVneu和Pten398A/398A;MMTVneu小鼠的石蜡包埋肿瘤标本进行免疫组化检测γH2AX。采用半定量方法对γH2AX免疫反应性进行评分(表1)。Pten+/+;MMTVneu肿瘤一般很少表达γH2AX(图1B)。所有Pten398A/398A;MMTVneu样本均为γH2AX强阳性,表明更高水平的基因组不稳定性(图1B, C)。我们通过对肿瘤样本的western blot分析证实了这些结果,结果显示Pten398A/398A;MMTVneu肿瘤比Pten+/+;MMTVneu肿瘤表达更高水平的γH2AX(图1D, E)。通过对Ki-67进行免疫组化来评估肿瘤的增殖指数,Ki-67是常规病理中广泛使用的标志物。两种基因型Pten+/+肿瘤的Ki67免疫反应性无显著差异;MMTVneu(补充图2C, D)。总之,Pten398A突变加速mmtv神经驱动的肿瘤发生,这与更高的基因组不稳定性有关,但在细胞增殖中没有明显的改变。
二、阻断atm依赖的PTEN磷酸化导致基因组
为了确定ATM介导的PTEN磷酸化如何调节DDR,构建Pten398A/398A和Pten+/+ littermates MEFs细胞模型。PTEN蛋白水平不受398A突变的影响(补充图3A, B)。此外,磷酸化AKT (PI3K通路活性的标记物)水平在Pten398A/398A和PTEN +/+ MEFs中相似(补充图3B)。构建人乳腺上皮细胞系(MCF10A),稳定表达野生型PTEN (PTEN- wt),或PTEN的突变形式,不能被ATM磷酸化(PTEN- 398a)。在这些细胞中,内源性的PTEN基因被CRISPR/Cas9编辑删除,创建了PTEN空细胞,然后用野生型或突变型的蛋白质稳定转导重组。在表达PTEN- wt和PTEN- 398a的MCF10A细胞中,PTEN蛋白和磷酸化AKT水平相似(补充图3A),使这些细胞成为ATM磷酸化PTEN分子作用研究的合适替代模型。
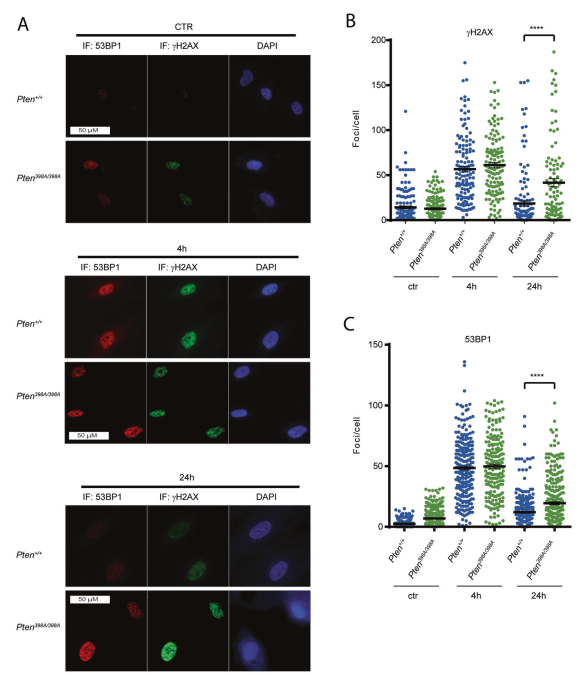
Pten+/+和Pten398A/398A mef在10gy IR作用下,通过测定每个细胞的γH2AX和53bp1病灶数量来评估其修复DNA损伤的能力。在Pten+/+ mef中,我们观察到在照射后4小时,每个细胞的病灶数量达到峰值,24小时后恢复到接近基线水平(图2A C)。Pten+/+和Pten398A/398A在ir后4小时,每个细胞聚集的病灶数量相似。然而,Pten398A/398A mef在24小时时间点比Pten+/+ mef保留了更多的病灶数,表明DNA修复能力降低(图2 C)。
三、pten - s38a突变诱导细胞凋亡抵抗
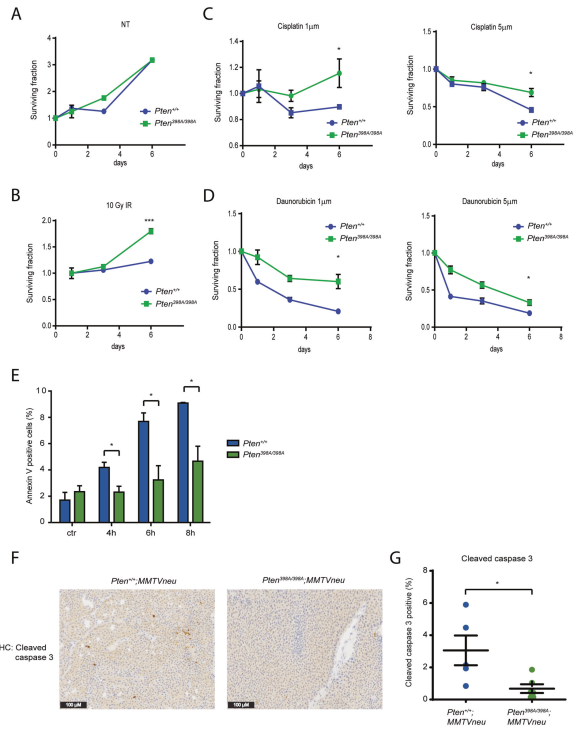
为了进一步了解阻断atm依赖的PTEN磷酸化如何影响细胞对基因毒性损伤的反应,我们使用抗体阵列评估了表达PTEN- wt或PTEN- 398a的MCF10A细胞裂解液的应激反应和凋亡通路的活性。Pten+/+和Pten398A/398A MEF培养,表达Pten - wt的MCF10A细胞与表达Pten -398A的MCF10A细胞在正常培养条件下的生长速率均无差异(图3A和补充图5C)。对细胞进行不同的基因毒性胁迫处理:IR、顺铂和柔红霉素。在所有条件下,Pten398A/398A MEFs和PTEN-398A表达的MCF10A细胞对基因毒性胁迫的抗性均高于野生型PTEN的细胞(图3B D和Supplementary Fig. 5D, E)。通过流式细胞仪Annexin V染色判断(图3E)。通过使用caspase-3抗体对Pten+/+、MMTVneu和Pten398A/398A、MMTVneu肿瘤进行免疫组化染色,证实了这些观察结果。caspase-3抗体是一种广泛用于检测凋亡细胞的标志物。确实,MMTVneu肿瘤的caspase-3阳性细胞比相应的Pten+/+少;综上所述,这些结果表明,PTEN不能被ATM磷酸化的细胞对基因毒性应激诱导的细胞凋亡具有抗性。为了评估表达PTEN-398A且DNA损伤未解决的细胞在辐照(图2B, C和补充图4B, C)后的持续存在是否是诱导凋亡反应失败的结果,我们在IR之前先用泛caspase抑制剂z-VAD-FMK (zVAD)对细胞进行预处理。与DMSO处理相比,zVAD增加了PTEN-WT细胞中未解决的DNA损伤灶的数量,其水平与DMSO处理的PTEN-398A细胞相当(补充图5F)。然而,需要注意的是,zVAD预处理也略微增加了IR后PTEN-398A细胞中未解决的DNA损伤灶的数量。
四、atm依赖性PTEN磷酸化缺失的细胞中G1/S细胞周期检查点受损
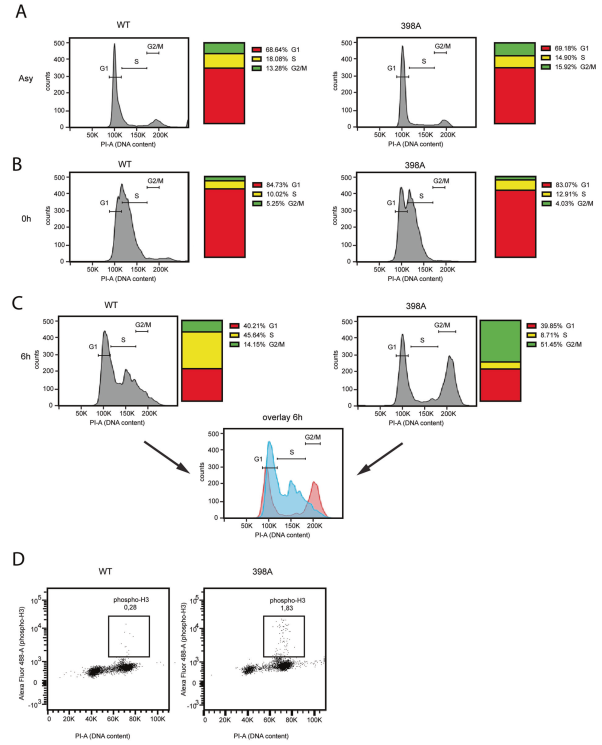
对表达PTEN-WT或PTEN-398A的MCF10A细胞进行了cDNA微阵列分析,并用顺铂诱导基因毒性应激。差异表达基因列表采用基因集合富集分析[39]进行分析。有趣的是,表达PTEN- 398A的细胞中上调的基因与G1/S检查点的激活有关,如G1/S期特异性转录、E2F靶标、Rb1通路控制的细胞周期调控基因等推测表达PTEN-398A的细胞对基因毒性胁迫的抗性可能是由于未能阻止细胞周期以应对DNA损伤。为了测试这种可能性,测量了表达野生型或突变PTEN的同步细胞在细胞周期阻滞在G1/ S检查点的能力,以应对DNA损伤。在表达PTEN-WT或PTEN-398A的正常周期细胞中,G1、S、G2/M期细胞比例没有明显差异(图4A)。为了使细胞在G1/S检查点同步,我们对它们施加双胸腺嘧啶脉冲。这种处理有效地抑制了PTEN-WT和PTEN-398A细胞在G1/S边界(图4B)。然后在正常生长培养基中释放细胞,在S期进展期间用IR处理,6小时后收集细胞,用流式细胞术分析细胞周期分布(实验方案见补充图7A)。大部分PTEN-WT细胞被阻滞在s期,而PTEN-398A细胞未能阻滞细胞周期,而是发展到G2/M期(图4C)。通过测定表达磷酸化组蛋白H3 (Ser10)的细胞比例,证实了这一观察结果。磷酸化组蛋白H3 (Ser10)标志着细胞处于有丝分裂阶段。(图4 d)。
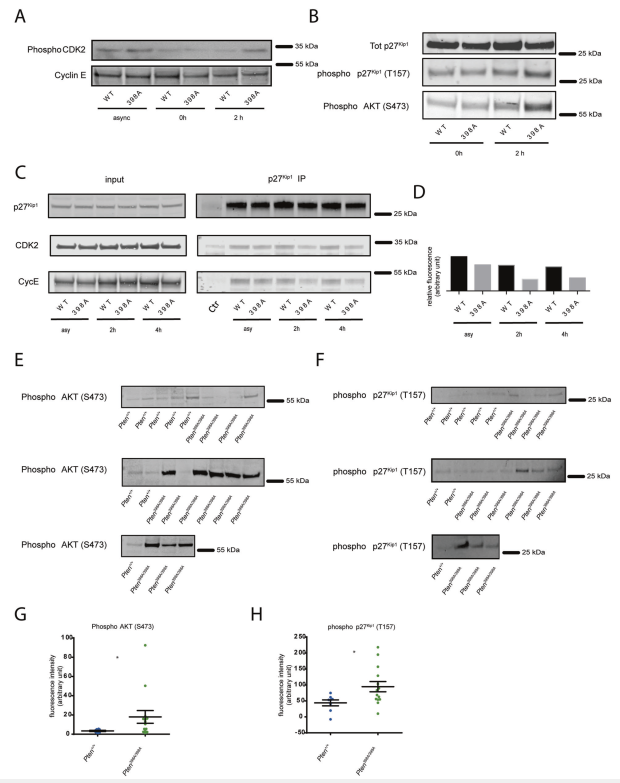
表达PTENWT的细胞,在双胸苷激酶阻断和辐射释放后被阻滞在S期,表现出低水平的CDK2磷酸化(图5A)。相反,经过相同处理的PTEN-398A细胞有更高水平的磷酸化CDK2(图5A),这与DNA损伤检查点的缺陷激活一致。与PTEN-WT细胞相比,PTEN-398细胞中磷酸化的AKT和磷酸化的p27Kip1水平更高(图5B)。虽然p27Kip1与CDK2和Cyclin E在照射过的PTEN-WT细胞中有效地共免疫沉淀,但在PTEN-398A细胞中这些相互作用减弱(图5C, D)。与我们在MCF10A细胞中观察到的结果一致,免疫印迹分析显示PTEN398A/398A裂解物中磷酸化的AKT和磷酸化的p27Kip1水平更高;MMTVNeu肿瘤与Pten+/+;MMTVNeu对应肿瘤进行比较(图5E-H)。总之,抑制ATM对PTEN的磷酸化增加了DNA损伤后AKT和p27Kip1的激活,反过来有利于CDK2/Cyclin E复合物的活性,并驱动细胞周期进程。
五、阻断atm依赖的磷酸化会损害DNA损伤后PTEN的亚细胞再分配
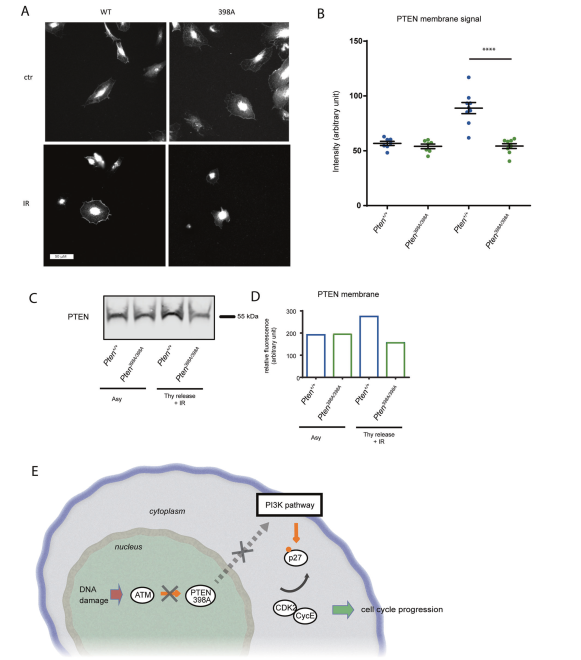
研究了磷脂酰肌醇3,4,5磷酸(PIP3)在DNA损伤后的细胞定位。将MCF10A细胞按上述方法同步辐照,免疫荧光法分析PTEN的亚细胞分布。PTEN-WT和PTEN-398A蛋白在细胞核和质膜稳定定位(图6A, B)。经过IR处理后,PTEN-WT在质膜上积累(图6A, B)。相比之下,在这些条件下,没有检测到PTEN-398A蛋白的重新定位(图6A, B)。B).我们通过细胞分离和膜相关PTEN的免疫印迹分析进一步证实了这些结果(图6C, D)。这些结果表明,ATM对PTEN的磷酸化导致PTEN在质膜上重新分布,这与AKT通路激活减少有关。