






circPDE4B-关节炎的靶点
circRNAs已经成为重要的生物调节因子。小编今天给大家带来2021年5月发表于影响因子为16.102的Ann Rheum Dis杂志上题为"circPDE4B prevents articular cartilage degeneration and promotes repair by acting as a scaffold for RIC8A and MID1"的文章。在本研究中,作者旨在阐明circPDE4B的作用,据报道,该circRNAs在骨关节炎(OA)组织中下调。我们体外研究了circPDE4B在人和小鼠软骨细胞中的作用。通过RNA下拉-质谱分析、免疫共沉淀、谷胱甘肽- S-转移酶下拉、RNA免疫共沉淀,验证了circPDE4B与RIC8A)/ MID1复合物的相互作用。采用小鼠OA模型证实了circPDE4B在体内OA发病机制中的作用。本研究强调了circPDE4B-RIC8A轴在OA关节中的功能及其对MAPK-p38的调控,提示这一轴可能是OA的治疗靶点。

技术路线
结果
1)circPDE4B在OA组织中表达较低
我们之前对3个临床OA和3个对照样本的软骨细胞核糖体RNA缺失的总RNA进行了RNA-seq分析。在数量最多的50个显著调控异常的circRNA中,circPDE4B的表达水平排名第一。我们评估了每个病例的OA严重程度(图1A)。同时,我们进行了组织形态学和荧光原位杂交实验,结果显示软骨降解程度增加对应软骨细胞中circPDE4B的表达降低(图1B)。RT- qPCR检测到严重OA组织软骨细胞中circPDE4B RNA水平下调,而mPDE4B mRNA水平保持相对一致(图1C)。综上所述,circPDE4B的表达与OA的严重程度呈负相关。
考虑到circPDE4B在人和小鼠之间是保守的,我们也检测了circPDE4B在人/小鼠软骨细胞中的表达,发现IL-1β和TNF-α处理显著降低了HCs(人)/MCs(鼠)中circPDE4的表达,且呈时间依赖性(图1D)。核分离实验结合RT- qPCR分析和FISH分析发现,circPDE4B/circPde4b主要位于HCs/MCs的细胞质中(图1E、F)。综上所述,circPDE4B在OA中被下调,因此可能参与了OA的进展。
我们接下来试图识别circPDE4B上游调节器。我们首先对circPDE4B侧翼序列进行RNA pull-down-MS检测,发现两个与RNA剪接相关的RBP,包括DExH-box helicase 9和FUS(图1G)。RT-qPCR结果显示,FUS敲除后,HCs中circPDE4B表达下调,而pPDE4B和mPDE4B没有明显变化(图1H)。此外,感染两种FUS shRNA慢病毒只降低了circPDE4B的表达(图1I),而过表达FUS则上调了circPDE4B的表达(图1I)。接下来,RIP分析显示FUS与外显子相邻的位点结合,而其他远端位点则可以忽略(图1J,K)。我们还寻找了可能的FUS响应元素,发现了两个可能的motifs,A位于上游,B位于下游。我们进一步设计了两个短的circPDE4B微基因,包括circPDE4B-s和circPDE4B-s-del(图1L)。RIP显示FUS与circPDE4B-s之间存在明显的相互作用,但与circPDE4B-s-del之间没有相互作用(图1M),表明FUS需要周围内含子中的假定位点来结合。我们接下来在表达circPDE4B-s/del的HCs中敲除FUS,发现与circPDE4B-del相比,circPDE4B-s显著减少了FUS敲除的circPDE4B转录本(图1N)。值得注意的是,在HCs中FUS被TNF-α下调(图1O)。综上所述,在OA中circPDE4B的下调至少部分是由FUS的抑制引起的。

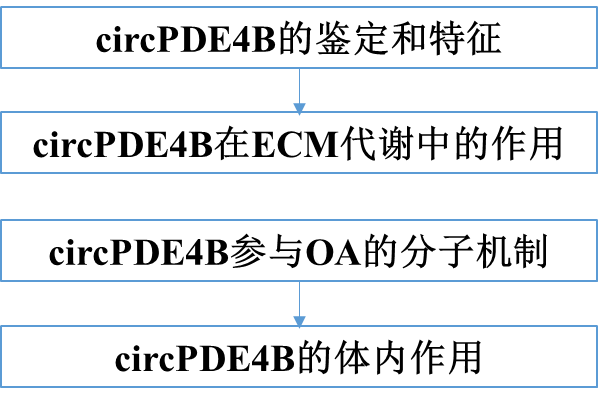
为了评估circPDE4B在ECM代谢中的作用,我们分别用三种circPDE4B siRNA转染HCs(图2A)。我们使用CCK-8方法评估了circPDE4B对软骨细胞活力的影响。结果显示,敲低circPDE4B的表达降低了软骨细胞的活力(图2B)。此外,shRNA腺病毒抑制circPDE4B显著提高了MMP3、MMP13和ADAMTS4的表达,而SOX9、COL2A1(或COL2蛋白)和aggrecan在HCs/MCs中的表达下调(图2C和图2D)。免疫荧光进一步证实,circPDE4B敲除影响了HCs/MCs的MMP3、MMP13、COL2和aggrecan水平(图2E、F)。同时,HCs的阿尔新蓝染色显示,circPDE4B抑制导致软骨细胞功能障碍,蛋白多糖蓝染较少(图2G)。然后通过CCK-8实验(图2H)发现,过表达circPDE4B增加了软骨细胞的活力。在过表达circPDE4B -HCs中,MMP3、MMP13和ADAMTS4的mRNA和蛋白水平下调,SOX9、COL2A1(或COL2蛋白)和aggrecan的mRNA和蛋白水平上调(图2I-L)。此外,HCs的阿尔新蓝染色表明,与单独IL-1β治疗相比,circPDE4B过表达和IL-1β共处理减少了软骨破坏(图2M)。这些数据表明,circPDE4B/ circPde4b在HCs中可以促进细胞活力,抑制分解代谢作用。
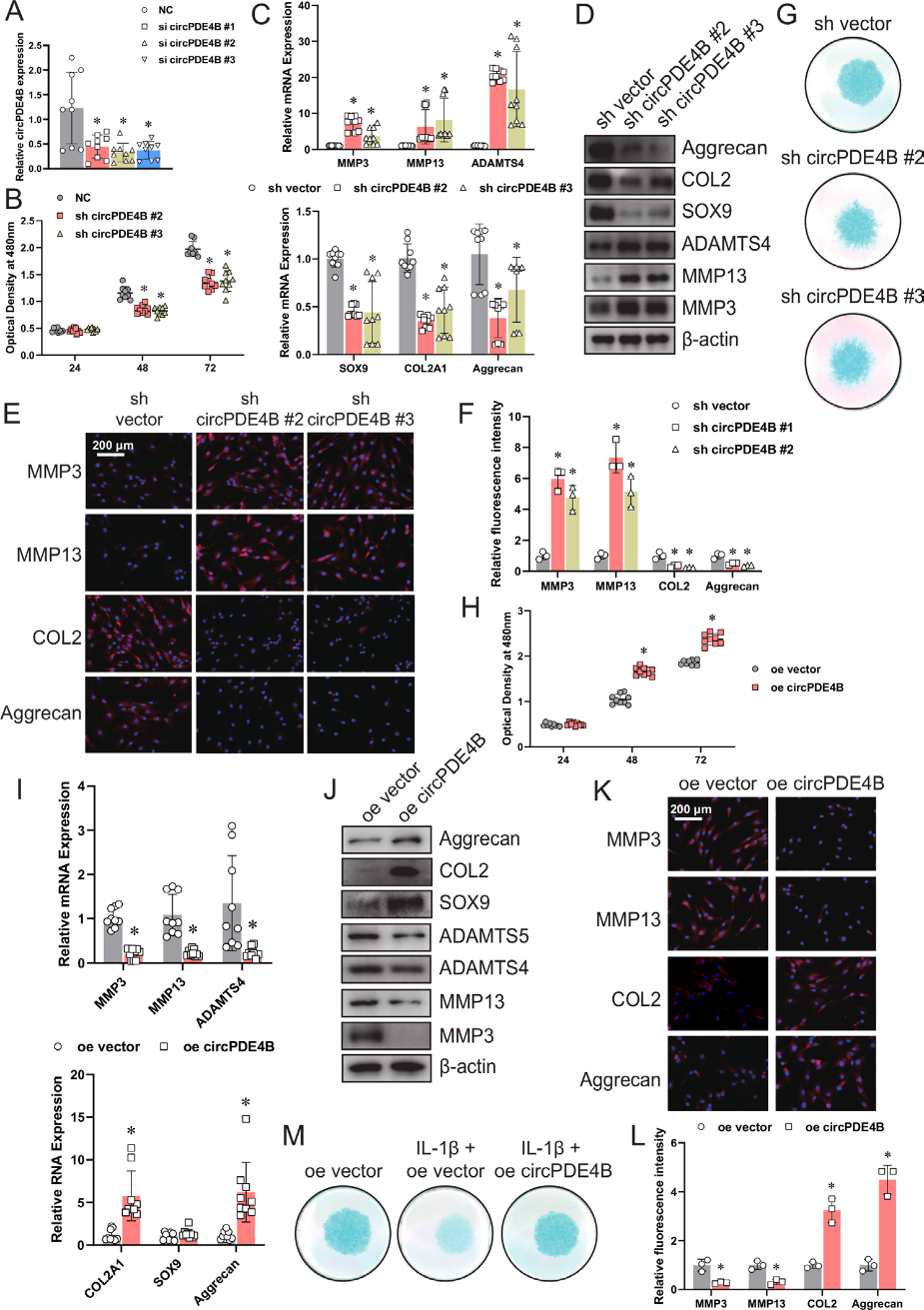
为了鉴别与circPDE4B相互作用的蛋白,我们采用RPD-MS(图3A),共鉴定出112个与circPDE4B相互作用的蛋白(图3B),确定了RIC8A。HCs中的RNA-蛋白共定位也证实了RIC8A和circPDE4B之间的相互作用(图3C)。RPD实验表明,体外线性转录的circPDE4B能够拉下重组RIC8A(图3D)。然后,我们使用catRAPID工具预测circPDE4B和RIC8A的相互作用区域(图3E)。为了识别预测的结合位点,我们将FL circPDE4B截短为3个片段。根据预测,RIP结果显示只有FL和S3被RIC8A拉下(图3F)。有综合来看,这些结果表明circPDE4B在HCs中与RIC8A相互作用。我们进一步的研究表明,circPDE4B调控RIC8A蛋白水平,而不是mRNA水平或稳定性(图3G-I)。我们还阻断了RIC8A蛋白的合成,并观察到sh-阴性对照(NC)和sh-circPDE4B 的HCs之间的RIC8A蛋白半衰期有明显差异(图3J),说明circPDE4B降低了RIC8A蛋白的稳定性。经PS341处理后,RIC8A在circPDE4B过表达和下调细胞中均未发生变化(图3K,L),表明circPDE4B通过蛋白酶体活性调节RIC8A。无论内源性还是外源性RIC8A, RIC8A的多泛素化在circPDE4B耗尽后均下降,在过表达circPDE4B后则上升(图3O)。综上所述,circPDE4B翻译后影响蛋白酶体介导的RIC8A的降解和周转。

4)circPDE4B促进了RIC8A和MID1之间的三元配合物的形成,促进了RIC8A的降解
我们试图鉴定参与RIC8A蛋白酶体降解的E3连接酶。有趣的是,MS结果显示circPDE4B也结合了两个E3连接酶,包括RNF2和MID1。然而,由于RNF2定位于细胞核内,我们选择MID1进行进一步研究。实际上,通过免疫沉淀分析,MID1被发现与RIC8A结合(图4A)。RIC8A和MID1的免疫荧光染色也证实了它们在HCs中共定位(图4B)。IP结果表明,MID1敲低有效地削弱了RIC8A和MID1过表达的泛素化作用,增加了RIC8A泛素化作用(图4C)。Co- IP实验也显示,与对照组相比,circPDE4B敲低细胞中RIC8A和MID1的结合减少,而过表达circPDE4B则有相反的作用(图4D)。此外,circPDE4B不影响MID1水平(图4E)。RPD和序列IP分析都显示circPDE4B促进了RIC8A和MID1的结合(图4F,G)。与这一发现相一致的是,体外结合试验表明,circPDE4B增加了重组RIC8A和MID1蛋白之间的关联(图4H)。通过co-IP,MID1与RIC8A在N端调控域结合(图4I)。此外,我们检测了RIC8A的功能位点。在HCs中免疫沉淀RIC8A并进行MS分析,证实了RIC8A中氨基酸残基的泛素化(图4J)。在RIC8A、K143和K187中鉴定出10个泛素化位点,并且在人类和小鼠之间并不保守(图4J)。因此,我们将保守的RIC8A位点从赖氨酸(K)突变为精氨酸(R),以排除泛素化。IP结果表明,与WT相比,K415的取代大大降低了RIC8A的泛素化位点(图4K)。有趣的是,K415在哺乳动物中高度保守(图4L,M)。此外,在K415突变后,circPDE4B过表达或抑制不再调节RIC8A的泛素化水平(图4N)。这些结果表明circPDE4B可以作为促进RIC8A和MID1之间联系的支架。

5)circPDE4B和RIC8A调控软骨细胞中的p38信号通路
为了阐明RIC8A下游的信号通路,我们研究了RIC8A敲低的HCs中MAPKs、NF-κ B和mTOR的磷酸化水平。两种RIC8A shRNA显著降低了p38的磷酸化水平(图5A)。然后用信号分子抑制剂对HCs进行预处理,包括ERK1/2抑制剂、p38抑制剂和JNK抑制剂,然后是RIC8A过表达。经过p38 MAPK抑制剂预处理的RIC8A过表达抑制了OA,然而,ERK或JNK抑制剂对其没有影响(图5B)。此外,感染RIC8A shRNA或过表达腺病毒后,p38 MAPK的磷酸化及其定位发生了异常调节(图5C-E)。这些结果表明,RIC8A在软骨细胞中通过p38信号通路发挥作用。接下来我们研究了circPDE4B在OA中调控p38信号通路中的作用。circPDE4B过表达降低,而circPDE4B敲低激活p38 MAPK信号,同时p38磷酸化和核转位(图5F-H)。然后我们进行了拯救试验。如图5I-K所示,RIC8A过表达挽救了过表达circPDE4B诱导的p38信号通路的下调,而抑制RIC8A挽救了敲除circPDE4B诱导的p38信号通路的激活,以及p38的磷酸化和核易位。基于这些发现,circPDE4B-RIC8A轴在调节软骨细胞下游p38 MAPK信号通路中发挥重要作用。

6)circPde4b和RIC8A影响小鼠OA发病机制
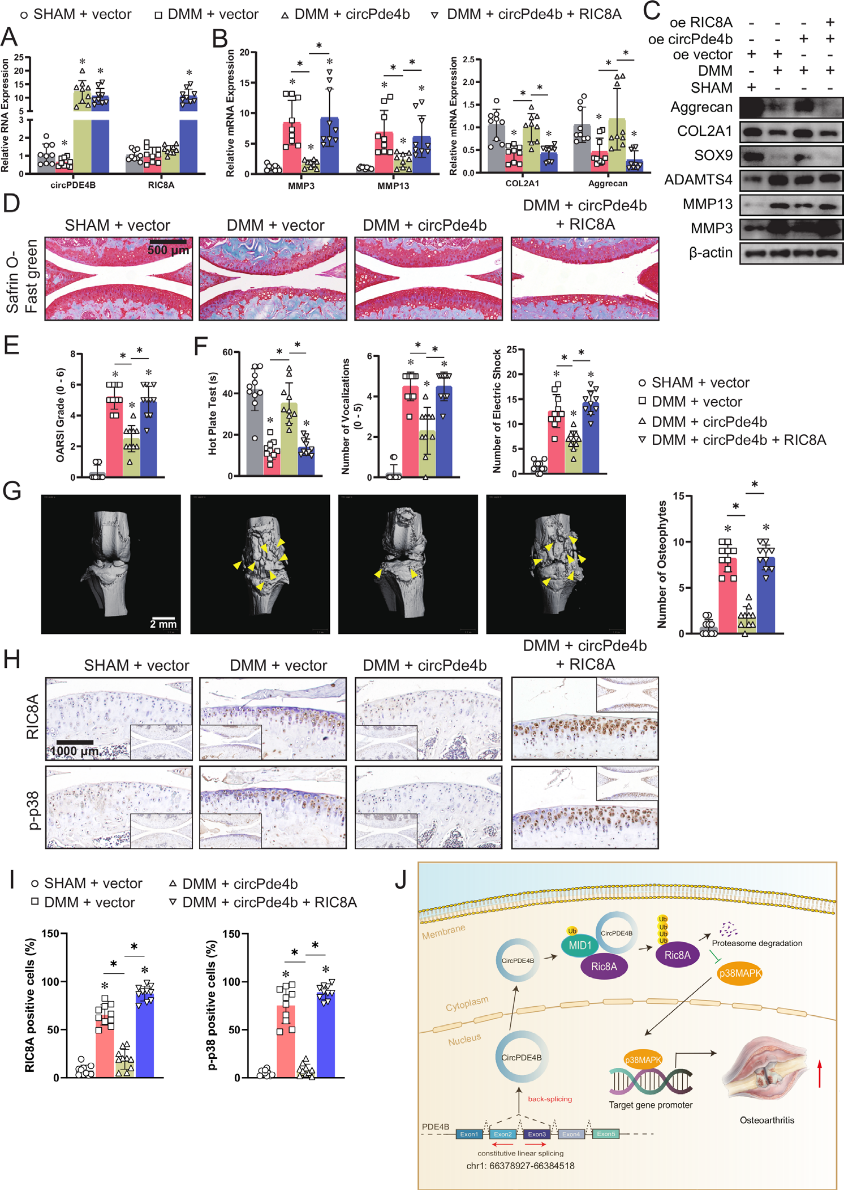
为了证实上述发现,我们进一步评估了circPde4b对小鼠OA的影响。图6A为四组不同AAV感染后circPde4b和RIC8A的RNA表达情况。从软骨中提取的ECM相关蛋白的RT-qPCR和western blot分析也提示在内侧半月板不稳(DMM)+vector组和DMM+ circPde4b+RIC8A组中OA更严重(图6B,C)。Safranin O fast green染色(图6D)发现,与SHAM+载体组和DMM+circPde4b+RIC8A组相比,DMM+载体组和DMM+circPde4b+RIC8A组的蛋白多糖明显丢失,表明circPde4b AAV可以挽救由DMM引起的OA进展,而RIC8A AAV可以逆转这种挽救。OARSI分级(图6E)进一步提示小鼠SHAM+载体和DMM+circPde4b组软骨降解较少,而DMM+载体和DMM+circPde4b+RIC8A组软骨降解情况相反。热板试验、膝关节伸展试验和电击刺激跑步机试验显示,DMM+载体组和DMM+circPde4b+RIC8A组的不适和膝关节疼痛多于SHAM+载体组和DMM+circPde4b组(图6F)。小鼠膝关节的三维微CT重建显示,DMM+NC组和DMM+circPde4b+RIC8A组比SHAM+载体和DMM+circPde4b组有更多的骨赘(图6G)。此外,四组中RIC8A和p-p38标记的免疫组化染色显示过表达circPde4b下调了RIC8A和p-p38的表达,从而抑制了DMM手术引起的OA进展(图6H,I)。综上所述,在小鼠中,circPde4b和RIC8A参与了OA的发病机制(图6J)。
结论:我们的研究描述了一种新的circRNA在OA中的机制。我们发现circPDE4B可以作为蛋白质降解的支架,在OA的进展中发挥重要作用。在临床前动物模型中,CircPDE4B被发现调节细胞外基质代谢,阻止软骨基质的形成,证实了其对OA发生的潜在治疗作用。机制上,circPDE4B可作为促进RIC8A-MID1结合的支架,降低RIC8A依赖的p38信号通路的激活,从而调节OA的进展。