






RAS GTPase RIT1通过抑制纺锤体组装检查点降低有丝分裂的保真度

2021年6月,最新一篇发表在Curr Biol(IF=9.601)的文章(The RAS GTPase RIT1 compromises mitotic fidelity through spindle assembly checkpoint suppression.)从共生功能体视点的角度对29名接受同种异体造血干细胞移植的儿童的肠道、口腔和鼻腔微生物群进行了综合宿主微生物群分析。首次揭示了RIT1直接与SAC核心组件MAD2和p31conmet相互作用;CDK1在有丝分裂过程中磷酸化RIT1并抑制其与SAC的相互作用;RIT1是细胞有丝分裂及时进展所必需的;RIT1致病水平促进染色体分离错误。
背景:RIT1突变已被确定为肺腺癌的致癌因素和努南综合征的病因因素。RIT1有一组独特的效应蛋白,但与其他Ras GTPases共享MAPK通路的激活。然而,由于缺乏确定的同源GTPase激活蛋白或交换因子,RIT1 GTPase周期的调控仍不清楚。尽管如此,RIT1的丰度和活性是通过蛋白酶体降解在蛋白水平上调控的,这种机制是由适配器蛋白LZTR1和E3泛素连接酶Cullin 3 (CRL3LZTR1)介导的。RIT1在Noonan综合征中的作用很可能是通过MAPK通路的过度激活介导的,MAPK通路是该疾病的一种症状体征,但它在正常细胞和恶性肿瘤中的作用尚不清楚.
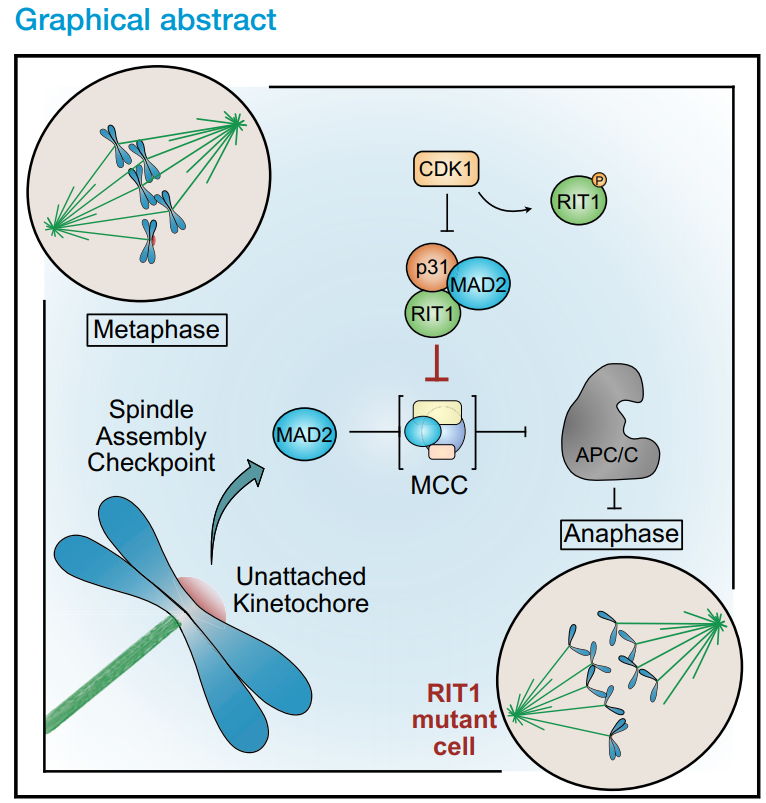
纺锤体装配检查点(SAC)作为一个传感器的独立的着丝点延迟有丝分裂进入后期,直到适当的染色体分离得到保证。这一安全机制的破坏导致基因组不稳定和非整倍体,这是胚胎死亡、先天性出生缺陷、智力残疾和癌症的遗传原因。然而,尽管了解了控制SAC的基本机制,但信号通路如何直接与有丝分裂检查点活动相互作用和调节仍是未知的。在对细胞外刺激的反应中,参与细胞生长、生存和分化的多种信号通路网络被激活,这一过程显著受到Ras家族小鸟苷三磷酸酶(GTPases)的调控。RIT1是一种ras相关的GTPase,调节细胞存活和应激反应,是通过有丝分裂和适当的染色体分离及时进展的必要条件。RIT1在有丝分裂期间从质膜(PM)分离,并直接与SAC蛋白MAD2和p31conmet相互作用,该过程受周期蛋白依赖性激酶1 (CDK1)活性的调节。此外,致病水平的RIT1沉默了SAC,并通过将MAD2从有丝分裂检查点复合体(MCC)中分离出来,加速了通过有丝分裂的转运。此外,致病性RIT1抑制SAC促进染色体分离错误和非整倍体。我们的研究结果突出了RIT1与其他Ras GTPases相比的独特功能,并阐明了信号通路与SAC之间通过一种新的调节机制的直接联系。
一、RIT1直接与SAC蛋白MAD2和p31conmet相互作用
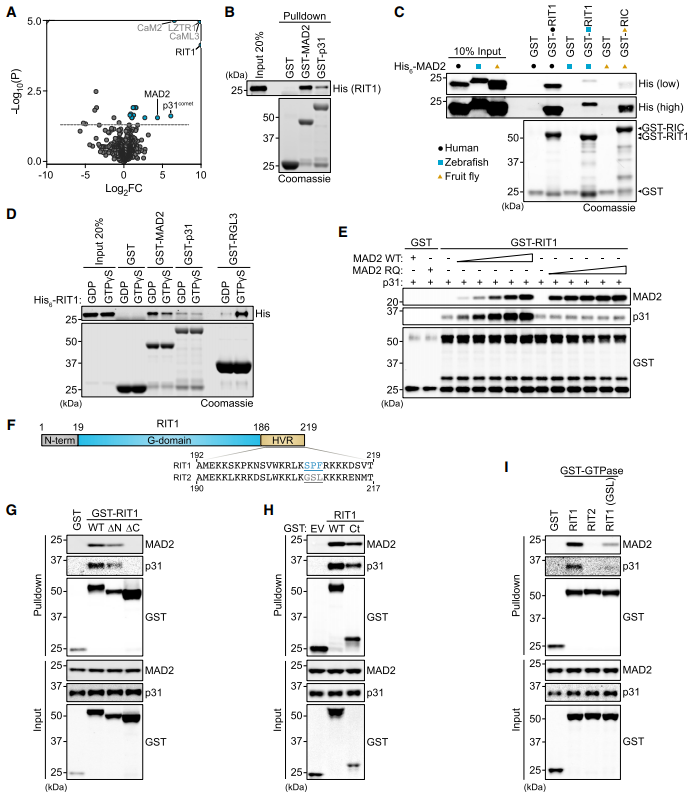
为了表征RIT1相互作用组,我们进行了亲和纯化-质谱筛选(图1A),确定MAD2 (MAD2L1)和p31conmet(也称为MAD2L1结合蛋白)作为新的和选择性的RIT1结合伙伴,不与其他Ras GTPases相互作用(图S1A和S1B)。
MAD2参与了在未附着的动点处催化MCC形成的SAC信号放大,MCC由MAD2、CDC20、BubR1和Bub3.1组成。16 MAD2和p31conmet二聚促使我们评估RIT1是否与MAD2和p31conmet直接相互作用。Pull down分析显示,这两种相互作用都直接且独立于MAD2和p31conmet二聚(图1B)。此外,RIT1-MAD2相互作用在斑马鱼和果蝇中是保守的(图1C)。为了确定RIT1与MAD2或p31conmet的结合是否受其GTPase周期的调控,我们评估了与GTPgS(一种不可水解的GTP类似物)加载的RIT1的结合。这表明这两种相互作用都不依赖于RIT1的鸟苷核苷酸负载状态(图1D)。一致地,与MAD2和p31conmet的结合不受与疾病相关的RIT1突变的影响(图S1C)。这些结果表明,结合界面位于对GDP/GTP结合敏感的RIT1 s开关I和II结构域外,因此MAD2和p31conmet不是典型的RIT1效应蛋白。MAD2和p31conmet的结构相似性突出了RIT1的潜在结合竞争。因此,我们使用了竞争结合试验,其中滴定MAD2野生型(WT)或R133E/Q134A (RQ),一个二聚体和p31结合缺陷的突变体,未能抑制rit1 -p31conmet结合(图1E)。由于RIT1 g结构域与其他Ras - GTPases,特别是它的邻域RIT2之间的相似性,假设RIT1 s的n端或c端延伸可能介导与MAD2和p31conmet的相互作用(图1F)。随后分析了RIT1 N-或c -末端缺失突变体,证明了c -末端结构域对于MAD2和p31conmet星结合是必要和充分的(图1G、1H和S1J)。
二、RIT1与MAD2和p31conmet的相互作用由CDK1磷酸化调控
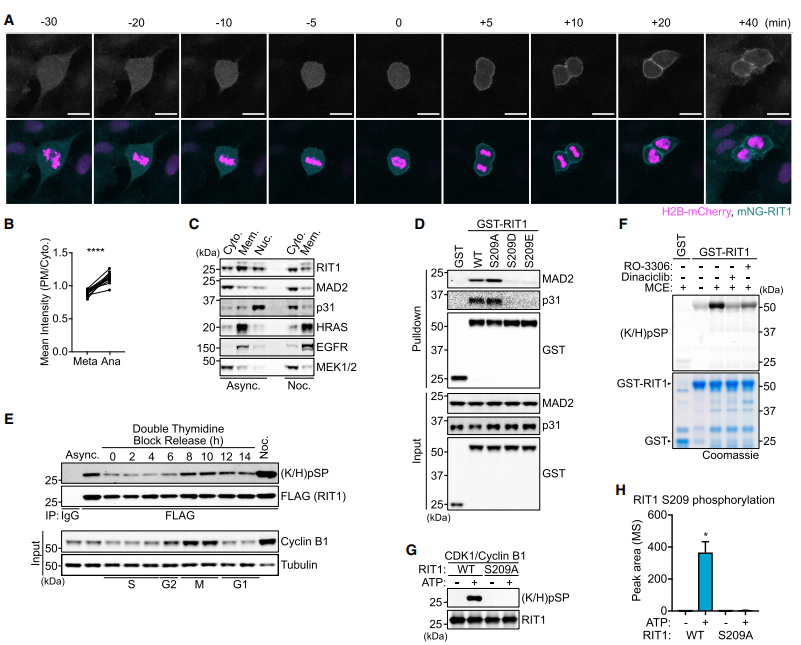
在间期,RIT1 s c端尾部介导质膜(PM)结合然而,在分析有丝分裂细胞时,我们观察到RIT1在细胞进入有丝分裂和进入中期并在后期快速易位到PM时的弥漫性胞质分布(图2A和2B)。同样,在有丝分裂细胞裂解物中检测到内源性RIT1的主要胞质分布(图2C)。(S209D/E)磷酸化,而非(S209A)缺磷,突变破坏了RIT1-MAD2/p31conmet结合(图2D)。RIT1磷酸化在有丝分裂过程中最为丰富(图2E)。抑制CDK1显著降低了重组RIT1的磷酸化(图2F)。免疫印迹检测和质谱证实CDK1/CyclinB1磷酸化RIT1 S209,(图2G和2H)。
三、RIT1及时调节后期进入和染色体分离保真度
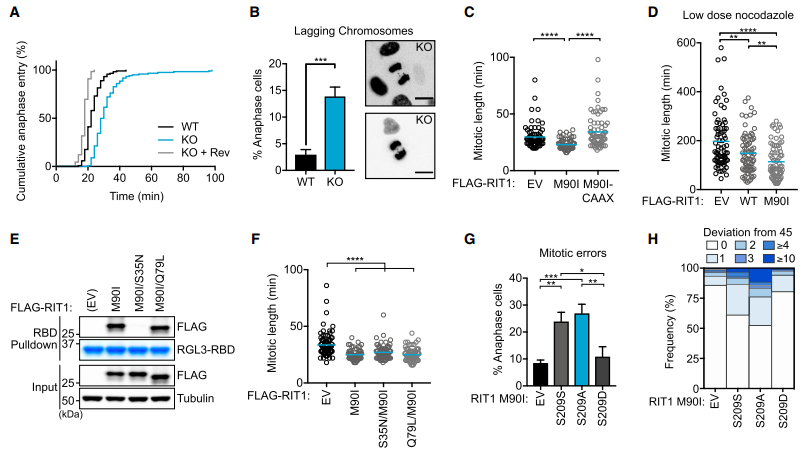
MAD2和p31conmet调节SAC的持续时间,反过来,也调节有丝分裂的持续时间。这促使我们研究RIT1是否通过与MAD2和p31conmet的直接关联来影响SAC。通过RNAi或crispr介导的敲除去除RIT1可延长有丝分裂进程(图3A和S3A S3E)。此外,SAC的药理抑制挽救了RIT1耗尽的作用,表明RIT1以SAC依赖的方式影响有丝分裂。此外,RIT1的缺失增加了染色体分离错误的发生率(图3B),这表明RIT1不仅对有丝分裂的及时进展至关重要,而且RIT1蛋白水平的失调也破坏了正常的SAC功能。LZTR1或RIT1M90I表达的缺失加速了异步生长细胞的有丝分裂进程,这一效应依赖于PM释放RIT1(图3C、S3H和S3I)。同样,RIT1 WT或M90I的过表达部分覆盖了药物诱导的SAC反应(图3D和S3J)。RIT1M90I的异位表达以依赖MAD2-和p31come结合的方式显著增加了有丝分裂错误的发生率,包括滞后染色体和桥接染色体(图3G和S3O)。因此,我们观察到在表达RIT1M90I的细胞中非整倍体率增加,但在表达不能结合MAD2/p31conmet的突变体的细胞中却没有(图3H和S3P)。这些结果表明,RIT1水平的增加会导致与MAD2和p31conmet的直接相互作用,从而降低有丝分裂的保真度。
四、RIT1抑制MCC-MAD2结合,促进APC/C底物降解
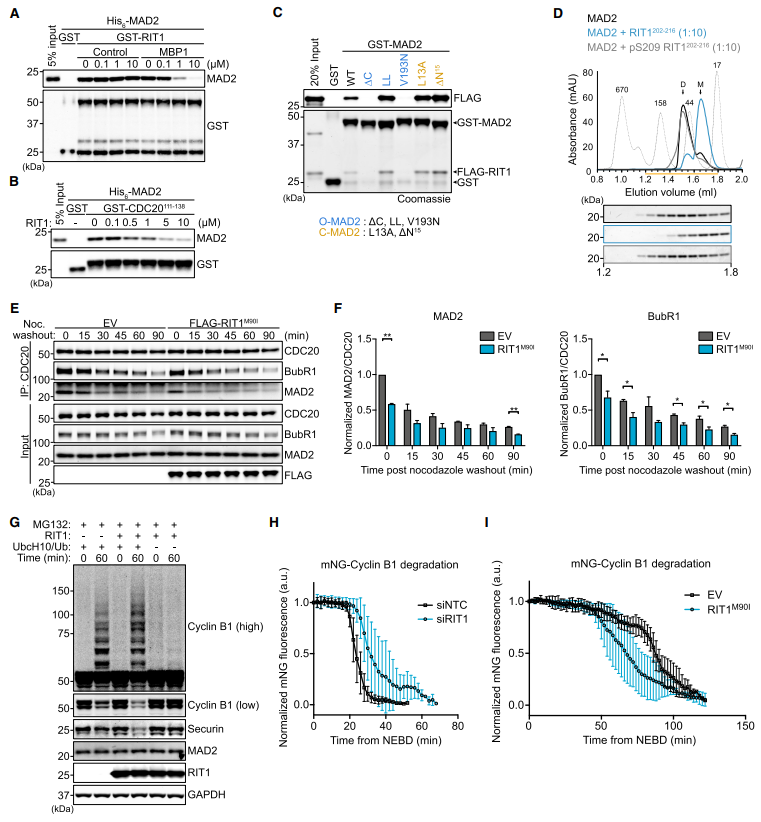
在MAD1与未连接的动点结合后,MAD2从开放的构象状态(O-MAD2)转化为封闭的构象状态(C-MAD2), SAC信号被紧密调控和放大。MAD2的构象变化启动了它与CDC20的结合。为了直接测试RIT1是否抑制了MAD2与CDC20或MAD1的关联,进行了竞争性pull-down实验来测试RIT1-MAD2和MAD2-CDC20/ MAD1结合之间的互斥性(图4A和4B)。MAD2结合肽1 (MBP1)是一种模拟CDC20和MAD1相互作用基序(MIM)的高亲和合成肽,它取消了MAD2- rit1的结合。评估了RIT1与O-或c -状态稳定的MAD2突变体的结合(图4C)。用过量的RIT1 c端尾肽孵育MAD2蛋白,并用凝胶过滤分离(图4D)。RIT1M90I显著降低了MAD2- CDC20和BubR1-CDC20的相互作用(图4E和4F);表明致病RIT1蛋白水平阻碍MCC的完整性,符合RIT1从CDC20中隔离MAD2并促进MCC拆卸的模型。
用重组RIT1补充这些提取物增加了CyclinB1和Securin的泛素化和降解,表明APC/C活性增加,可能是由于MCC抑制缓解(图4G)。RIT1缺失细胞显示CyclinB1降解延迟(图4H)。此外,RIT1M90I的表达加速了正常细胞生长下CyclinB1的降解(图4I);然而,其对CyclinB1降解的影响在药理学诱导的有丝分裂阻滞下被消除(图S4M),这表明致病性水平的RIT1不能抑制过度活跃的SAC反应。