






m6A阅读器YTHDC2通过抑制SLC7A11依赖的抗氧化功能来抑制肺腺癌的发生

m6A RNA甲基化是mRNA转录后最常见的内部甲基化。这是一个由m6A WER系统控制的可逆过程,由书写器(W)、擦除器(E)和阅读器(R)组成,而m6A甲基化的最终结果取决于特异性阅读器的识别并结合。目前研究发现抑制肺腺癌(LUAD)与m6A阅读器YTHDC2相关。该研究2021年1月发表在《Redox biology》,IF:11.799的期刊上。
技术路线:

1. YTHDC2表达在LUAD中被抑制,且与临床预后差相关
为了验证m6A阅读器在肺癌的表达,作者通过GEPIA数据库分析了LUAD中已知的阅读器,包括YTHDF1/2/3、YTHDC1/2、HNRNPA2B1、HNRNPC/G、eIF3A、FMR1和IGF2BP1/2。如图1A和B显示,与正常肺相比,LUAD 中YTHDC2、IGF2BP2表达下调,而结合Oncomine数据库和Kaplan-Meier图谱进一步分析,发现较低的YTHDC2与LUAD患者较差的总生存率相关(图1D),这证实了YTHDC2与LUAD存在负相关的关系。
接着作者为了进一步证明YTHDC2与LUAD的关系,与相邻的癌旁组织相比,在LUAD肿瘤中观察到YTHDC2 mRNA和蛋白的显著下调(图1E-G),组织芯片检测(TMA)评估cohort #1中YTHDC2的表达,结果显示51% (98/192) LUAD患者的YTHDC2表达下调(图1H、I)。值得注意的是,YTHDC2表达下调与更大的肿瘤直径和更晚期的分期相关(图1J、K)。表明抑制YTHDC2可促进LUAD的肿瘤进展。
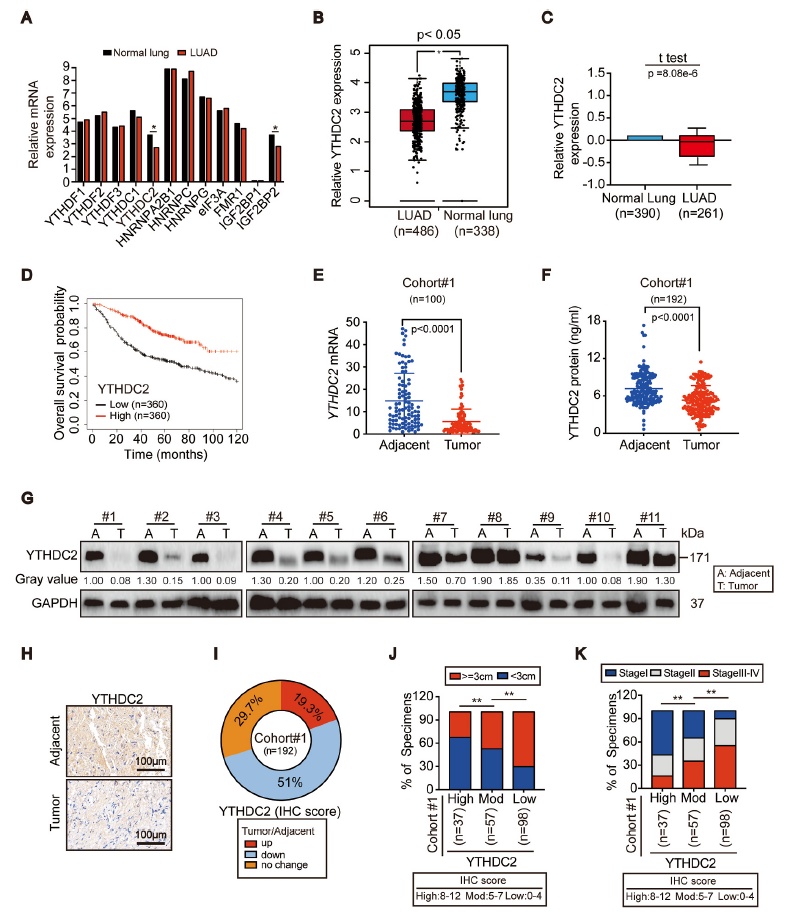
图1 YTHDC2表达在LUAD中被抑制,且与临床预后差相关
2. 过表达YTHDC2抑制细胞活力和肿瘤发生
为了探索YTHDC2在体内的m6A解读器功能,作者构建AAV5表达系统(KPYWT、KPYΔYTH和对照KPE)(图2A)。与KPE小鼠感染25周后的肿瘤相比,KPYWT和KPYΔYTH小鼠的肿瘤证实了YTHDC2持续上调(图2B),这表明AAV5表达系统具有持久的效率。虽然YTHDC2不能阻止肿瘤的发生,但肿瘤发生时间延迟,肿瘤负荷明显受到抑制,KPYWT小鼠的生存时间比KPE小鼠和KPYΔYTH小鼠更长(图2C-F)。
作者进一步研究YTHDC2是否是存在人类LUAD细胞中的一种肿瘤抑制因子。IB检测证实了YTHDC2的过表达和敲除效率(图2G和S2A)。然而在H1299细胞中过表达YTHDC2WT后,肿瘤3D球体的数量和大小以及异种移植瘤的生长下降,但在过表达YTHDC2ΔYTH时没有观察到这些现象(图2 H-K)。相比之下,敲除H1975细胞中的YTHDC2增加了小鼠的球体形成和异种移植瘤的生长(图S2B-E)。值得注意的是,当使用YTHDC2sg2−resistant (res)重组YTHDC2表达时,YTHDC2sg2在肿瘤发生中的阳性作用得到了恢复(图S2B-E)。因此,YTHDC2通过其m6A阅读域在LUAD中发挥肿瘤抑制功能。


图S2 敲除YTHDC2促进细胞活力和肿瘤发生
为了研究YTHDC2对代谢物的影响,作者对YTHDC2低表达和高表达的LUAD标本(分别称为YTHDC2low和YTHDC2high)进行代谢组学分析(n = 20/组)。如图3A显示,GSH代谢在KEGG通路中排名前20。在显著改变的代谢产物中,与YTHDC2high的肿瘤相比,YTHDC2low的肿瘤中胱氨酸含量增加了3.975倍(图3B)。此外,在cohort #1 (n = 100)中,YTHDC2和胞内胱氨酸呈负相关(图3C)。这些数据表明YTHDC2减少了细胞内的胱氨酸。
L-14c-胱氨酸摄取试验结果显示,YTHDC2通过其YTH域抑制了H1299细胞产生的异种移植物、小鼠形成的LUAD和患者来源的LUAD细胞的胱氨酸摄取(图3D-F)。图3H证明了系统Xc功能受损与GSH水平降低相关。NAC和GSH给药后,KPYWT小鼠的肿瘤负荷明显高于给药对照小鼠,且水平与KPE小鼠相似(图3H、I)。与KPE小鼠相比,GSH和NAC给药仅导致KPYWT小鼠肿瘤中GSH/GSSG比值显著增加。NAC和GSH的补充降低了KPYWT小鼠的脂质过氧化并缩短了存活时间(图3K、L)。

图3 YTHDC2抑制LUAD的胱氨酸摄取和下游抗氧化程序
4. YTHDC2抑制依赖于SLC7A11的胱氨酸摄取
Xc-系统是一个胱氨酸/谷氨酸逆向转运蛋白,捕获细胞外的胱氨酸,用于谷胱甘肽的合成,以交换谷氨酸的释放。在图3中,被抑制的Xc-系统功能与YTHDC2受损的胱氨酸摄取有关,然而YTHDC2如何调节Xc-系统依然未知。文献报道催化亚基SLC7A11在维持Xc-系统活性方面起着重要作用。作者通过IB 和RT-qPCR发现,SLC7A11蛋白和mRNA水平均受YTHDC2负调控(图4A、B)。在小鼠中,与KPE和KPYΔYTH小鼠相比,KPYWT小鼠的肿瘤中SLC7A11表达下调(图4C、D)。这证明YTH结构域是YTHDC2抑制SLC7A11表达的前提。
随后,作者研究了YTHDC2是否通过SLC7A11抑制胱氨酸摄取。当YTHDC2在H1299细胞中过表达时,并过表达SLC7A11完全恢复了受损的胱氨酸摄取(图4E-G)。ATF4是SLC7A11转录的关键转录因子,ATF4在H1299细胞中上调SLC7A11(图4H、I)。过表达YTHDC2仍然拮抗ATF4的作用(图4H-J)。因此,YTHDC2损害依赖于SLC7A11的胱氨酸摄取。结合临床分析,YTHDC2表达下调,而SLC7A11表达上调(图4K、L),并且它们在肿瘤中呈负相关(图4M)。

图4 YTHDC2抑制依赖于SLC7A11的胱氨酸摄取
5. YTHDC2以m6A依赖的方式使SLC7A11 mRNA不稳定
作者为研究YTHDC2是否在转录被阻断后调控SLC7A11 mRNA的稳定性,通过泛转录抑制剂ActD处理H1299和H1975细胞,发现YTH结构域对于YTHDC2缩短H1299细胞中SLC7A11 mRNA的半衰期至关重要(图5A)。在H1975细胞中,CRISPR/Cas9技术使YTHDC2功能丧失,然后YTHDC2重构,进一步证实YTHDC2加速了SLC7A11 mRNA的衰变(图5B)。EXOSC10是一种外切酶,负责3’-5’外切酶活性。EXOSC10的缺失,减弱了YTHDC2缩短SLC7A11 mRNA半衰期的作用(图5C、D)。在药理学水平上,用两种泛-RNase抑制剂DEPC和RNasin治疗H1299细胞,也阻断了YTHDC2的功能(图5D)。
YTHDC2调节SLC7A11 mRNA的稳定性(图5A-D),而控制RNA衰减是m6A甲基化的重要功能之一。作者先验证METTL3刺激m6A的能力(图5E、F)。在H1975细胞中敲降METTL3延长了SLC7A11 mRNA的半衰期,而在H1299细胞中过表达METTL3则加速了SLC7A11 mRNA的降解(图5G)。此外,H1299细胞中降低METTL3表达阻断了YTHDC2,从而减缓SLC7A11 mRNA的衰减,刺激胱氨酸摄取并减弱脂质活性氧(ROS)的生成(图5H-J),这表明YTHDC2在LUAD细胞中的作用是依赖m6A。
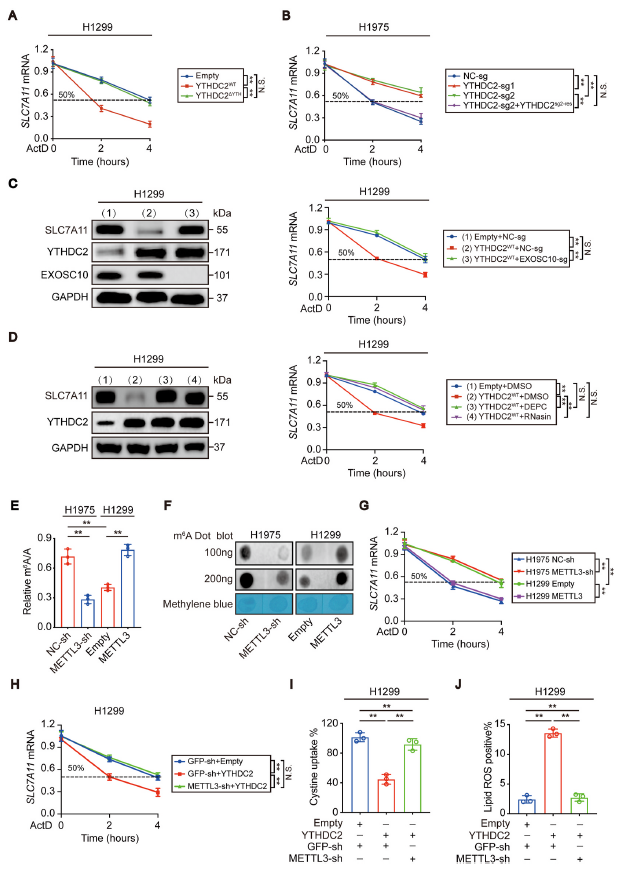
图5 YTHDC2以m6A依赖的方式使SLC7A11 mRNA不稳定
6. YTHDC2倾向于相互作用并使m6A甲基化的SLC7A11 mRNA不稳定
为了揭示SLC7A11 mRNA中潜在的m6A甲基化位点,在H1975细胞中METTL3敲除前后分别进行了MeRIP-seq研究。在H1975细胞中,无论是否敲除METTL3, GGAC motif都高度富集于m6A位点(图6A)。m6A峰在mRNA停止密码子附近的3’非翻译区(3’UTR)尤为丰富(图6B)。为了进一步证实SLC7A11 mRNA的m6A依赖性修饰,我们进行MeRIP-qPCR。在H1975细胞中,敲除METTL3后,SLC7A11 mRNA中的m6A修饰明显减少,特别是在假定的m6A位点周围。相反,当METTL3在H1299细胞中过表达时,观察到相反的结果(图6C)。
作者进行了PAR-CLIP实验,以评估SLC7A11 mRNA-YTHDC2相互作用是否依赖于m6A甲基化。结果表明,在H1299细胞中,通过过表达METTL3来提高m6A的甲基化水平,促进YTHDC2与SLC7A11 mRNA的结合(图6D)。无论METTL3是否过表达,YTH结构域的缺失都阻断了SLC7A11 mRNA-YTHDC2的相互作用(图6D)。作者通过SLC7A11 3’-UTR探针进行RNA-pull down实验,发现YTHDC2WT,优先结合于含有m6A的SLC7A11 mRNA的3'UTR,而不是含有未甲基化腺苷的mRNA(图6E)。此外,YTHDC2倾向于结合m6A共识基元GGAC (图6E)。通过在H1299和H1975细胞中进行METTL3的功能增加和丢失实验,发现细胞内SLC7A11 mRNA-YTHDC2的相互作用是由m6A水平决定的(图6F和G)。这些数据表明YTHDC2优先与m6A甲基化的SLC7A11 mRNA结合。
作者构建了 (WT) SLC7A113’-UTR和突变型(Mut) 3’-UTR两种质粒(图6H)。结果表明,只有过表达YTHDC2WT降低SLC7A11 mRNA在H1299细胞中的稳定性,敲除YTHDC2可以提高SLC7A11 mRNA在H1975细胞中的稳定性。然而,与WT 3’-UTR相比,Mut报告基因的这些作用减弱(图6I和J)。在检测外源性F-Luc-SLC7A11融合mRNA的RNA稳定性后,得到了类似的结果(图6K-M)。总的来说,3’-UTR上的m6A位点对于YTHDC2使SLC7A11 mRNA不稳定至关重要。

图6 YTHDC2倾向于相互作用并使m6A甲基化的SLC7A11 mRNA不稳定
7. 抑制YTHDC2对XC-系统靶向治疗敏感,并与LUAD进展相关
为了评估YTHDC2抑制在LUAD中的重要性,从cohort #1随机抽取20例YTHDC2low LUAD,其中50%属于腺泡型(图7A)。在cohort #2中,与相邻组织相比,肿瘤中YTHDC2表达下调,62%(62/100)的腺泡组织被归类为YTHDC2low(图7B、C)。与无这种表达模式的腺泡性LUAD患者相比,YTHDC2low的患者总生存期要短得多(图7D),进一步表明YTHDC2抑制可能对腺泡性LUAD的肿瘤进展尤为重要。另外,相对于癌旁组织,肿瘤中腺泡LUAD组织中SLC7A11 mRNA和蛋白水平表达量都比较高(图7E、F)。
来自4个不同腺泡性LUAD患者的PDX模型被给予XC-系统抑制剂erastin (PKE)和多激酶抑制剂索拉非尼(sorafenib)。与DMSO治疗的对照组相比,PKE和sorafenib给药时观察到显著的肿瘤消退(图7G-J)。此外,PKE和sorafenib给药小鼠也导致肿瘤中MDA和4-HNE的显著增加(图7K和L),提示诱导的脂质过氧化可能是抑制腺泡LUADs肿瘤发生的结果。
图7M显示,与癌旁组织相比,癌症组织中MDA和YTHDC2表达量都降低,而SLC7A11 mRNA和蛋白水平都升高,证明在LUAD中抑制YTHDC2可通过SLC7A11表达上调阻止脂质过氧化。同样地,MDA和YTHDC2与癌症期数呈负相关,而SLC7A11与分期呈正相关(图7N),说明SLC7A11的升高可能是抑制YTHDC2促进LUAD进展的关键。
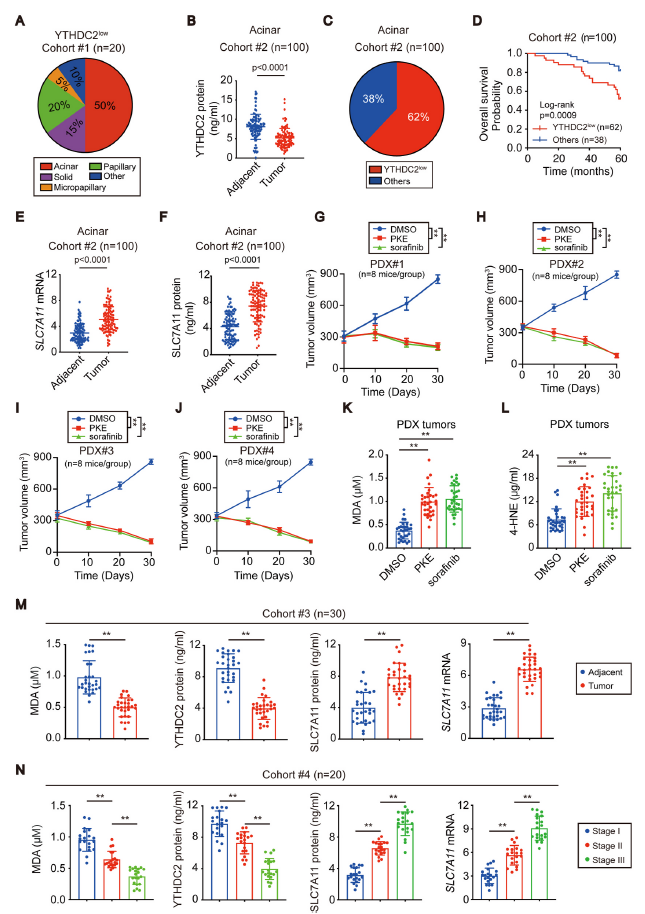
图7 抑制YTHDC2对XC-系统靶向治疗敏感,并与LUAD进展相关
结论:
本研究强调了m6A阅读器YTHDC2在LUAD中的重要性。XC−系统活性可能通过抑制YTHDC2来诱导促进肿瘤发生。此外,基于YTHDC2表达的分子模型可能有助于预测LUAD的预后。抑制剂靶向XC−系统可能有助于治疗预后不良的LUAD患者。因此,本研究对LUAD的肿瘤发生、分子分型和药物敏感性提供了有价值的见解。
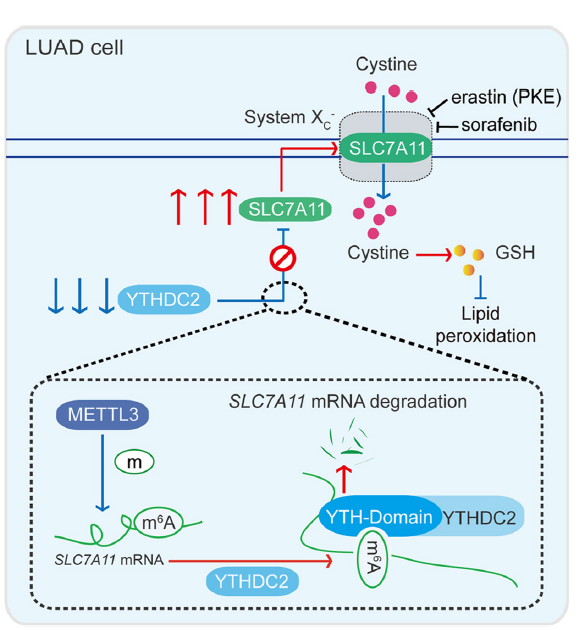
研究模型通路
参考文献:
Ma, L., Chen, T., Zhang, X., Miao, Y., Tian, X., Yu, K., Xu, X., Niu, Y., Guo, S., Zhang, C., Qiu, S., Qiao, Y., Fang, W., Du, L., Yu, Y., Wang, J. (2021). The m6A reader YTHDC2 inhibits lung adenocarcinoma tumorigenesis by suppressing SLC7A11-dependent antioxidant function. Redox Biol., 38:101801. doi:10.1016/j.redox.2020.101801