






在缺氧条件下持续刺激引起的线粒体应激快速驱动T细胞衰竭

2021年6月,最新一篇发表在Nat Immunol.(IF=25.600)的文章(Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion.)当肿瘤内衰竭T细胞经历严重缺氧时,他们假设代谢应激会改变其对其他信号的反应,特别是持续的抗原刺激。在体外,尽管单独经历连续刺激或缺氧的CD8 + T细胞分化为功能性效应物,但这种组合迅速驱动了与衰竭一致的T细胞功能障碍。连续刺激促进了Blimp-1介导的PGC-1α依赖性线粒体重编程阻遏作用,使细胞对缺氧的反应较差。线粒体功能的丧失产生了无法耐受的活性氧(ROS)水平,足以促进衰竭状态,部分原因是通过磷酸酶抑制和随后活化T细胞核因子的活性。减少T细胞固有的ROS和降低肿瘤缺氧限制了T细胞衰竭,与免疫疗法协同作用。因此,免疫信号和代谢信号之间存在内在联系:通过减轻代谢压力,可以改变T细胞分化,从而促进更多的功能性细胞命运。
背景:T细胞分化是一个复杂的过程,它整合了来自环境的大量信号,参与转录机制,并进行表观遗传改变来支持新的功能程序。对于CD8+ T细胞,这导致获得不同的命运:短命的细胞毒性效应程序和长寿命的自我更新记忆程序。然而,在抗原持续存在的病理中,特别是慢性感染和癌症中,会诱导一种替代效应或记忆分化的命运:T细胞衰竭。在持续的炎症提示下,T细胞逐渐失去多功能和更新能力,无法控制感染或恶性扩散。T细胞逐步上调共抑制分子,最终达到最终分化状态,表达高水平的PD-1和多种共抑制和共刺激标志物(lag3, Tim-3, TIGIT,和4 1BB)。衰竭的细胞由几种转录因子网络定义,包括Eomesodermin、BATF、无伴侣NFAT1、TOX和转录抑制因子Blimp-1,利用改变的表观遗传环境定义功能失调谱系。衰竭有一个不断演变的定义,而且只能在体内可靠地产生,这严重限制了识别这种命运的驱动因素的能力。
分化通常与T细胞接收的免疫信号有关:共刺激或细胞因子信号支持一种或另一种命运。然而,环境的代谢组成、检测该环境的营养传感器以及T细胞固有的代谢状态也对决定分化的最终结果至关重要。代谢信号是理解的关键,因为T细胞的激活和分化发生在各种代谢不同的组织环境。在癌症中,肿瘤细胞代谢的升高可以显著改变T细胞所经历的营养环境。我们之前的研究表明,T细胞浸润肿瘤时存在严重的代谢缺陷:葡萄糖摄取能力受到抑制,功能性线粒体质量丧失,伴随衰竭的发展。我们和其他人也表明,纠正这种错误的代谢状态(通过各种方法)可以增强免疫功能,实现免疫治疗效果。
例如,虽然之前认为抗PD-1可以阻断最末期枯竭的T细胞上的PD-1信号,但这种直接模型已经受到质疑,表明PD-1成功阻断可能作用于分化较低的类祖细胞,高比例的肿瘤浸润最终耗尽的T细胞预测抗pd -1的耐药。此外,虽然代谢相关因素,如肿瘤细胞消耗氧气和产生缺氧的能力,可以预测对PD-1阻断剂的抗性18,但这些环境压力源已被证明对T细胞功能有不同的影响。缺氧诱导因子1α (HIF1α)及其负调控因子此前已被证实与T细胞激活有关23,而在缺氧条件下治疗细胞的扩张可增强肿瘤杀伤能力。然而,无论是在隔离状态还是在体内,缺氧都具有明显的免疫抑制作用。因此,虽然代谢压力和T细胞衰竭有联系,但尚不清楚T细胞衰竭是否促进了导致细胞代谢改变的程序,或者是否代谢不足和压力直接导致了T细胞衰竭。
技术路线:

一、耗尽的肿瘤内T细胞会经历严重的缺氧
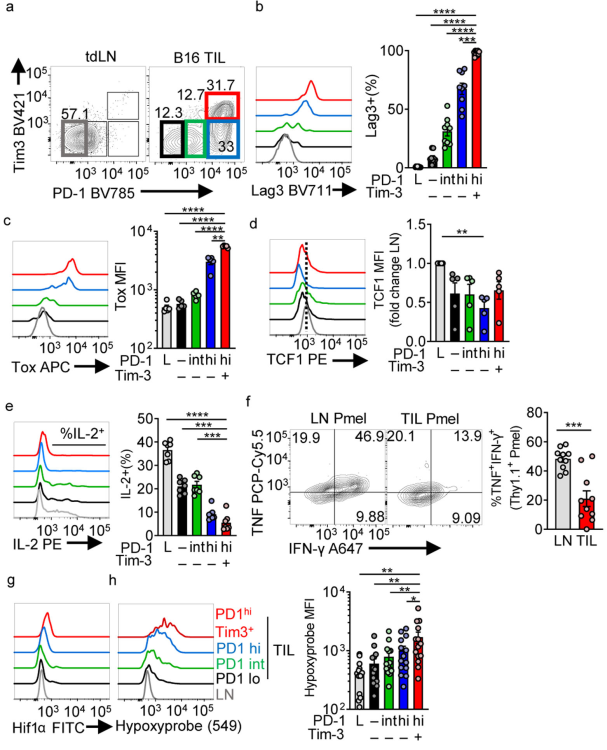
虽然缺氧在肿瘤中很常见,但尚不清楚肿瘤内T细胞亚群是否比其他T细胞更容易缺氧。我们首先在8-10 mm(第14天)B16黑色素瘤中沿着衰竭谱对CD8+肿瘤浸润淋巴细胞(TILs)进行表型分析(扩展数据图1a)。最终衰竭的T细胞被定义为高水平和持续表达PD-1和共表达Tim-3(图1a),具有高的LAG3和Tox表达,低的TCF1表达,通过将gp100特异的Pmel-1 T细胞转移到携带b16的小鼠中,一旦它们达到最终衰竭,就会重新刺激它们,从而测定抗原特异性反应(扩展数据图1b)。与LN-resident Pmel-1 T细胞相比,gp100-restimulated T细胞的多功能性要少得多(图1f)。在处死B16肿瘤小鼠之前,将其注入一种缺氧示踪剂吡莫硝唑,使用抗吡莫硝唑和抗hif1 α抗体,发现与其他亚群相比,最终耗尽的T细胞缺氧程度最高(图1g,h)。因此,缺氧是肿瘤代谢景观的主要代谢组成部分,与其他亚群相比,最终衰竭的T细胞会经历更高的缺氧。
二、在缺氧条件下持续刺激的T细胞出现衰竭

为了确定缺氧是否会导致细胞衰竭,在体外建立暴露于缺氧应激的模型。a,体外CD8+ T细胞d10的TNF和IFN-γ的产生,在缺氧或缺氧条件下,用抗cd3 /CD28珠急性或持续激活d2- 5,然后在常氧或缺氧条件下额外培养5天。b,,图2中使用的刺激方案生成的第3天CD8+ T细胞CTV稀释液的代表性直方图和division分析。c, CD8+ T细胞的扩增(左)和累积倍增(右)。d,第3天或第4天产生的CD8+ T细胞的染色稀释(增殖)与细胞死亡(活/死)染色。e, PD-1在cd4 + T细胞中的平均荧光强度。.f–h。第3天CD8+ T细胞的流式图。
三、在缺氧条件下的持续激活诱导不同的细胞内程序

已知的缺氧信号靶点BNIP3在缺氧下升高,mTORC1在持续刺激下被激活(图3a)。从这四种条件中获得的RNA序列被用来比较培养物和之前公布的数据之间的转录组。主成分分析显示,所有四个群体的转录组都不同(图3b)。缺氧条件下的持续刺激并没有诱导持续刺激和缺氧诱导基因的组合,而是驱动了一个不同的转录谱(图3c)。基因本体论表明,连续刺激条件之间共享的基因簇与负调控和代谢变化相关(簇3和簇5)(图3b)。与缺氧条件下持续刺激相关的基因(聚类1和7)主要由抗病毒和抗增殖基因驱动。
四、Blimp-1在持续刺激下抑制PGC1α
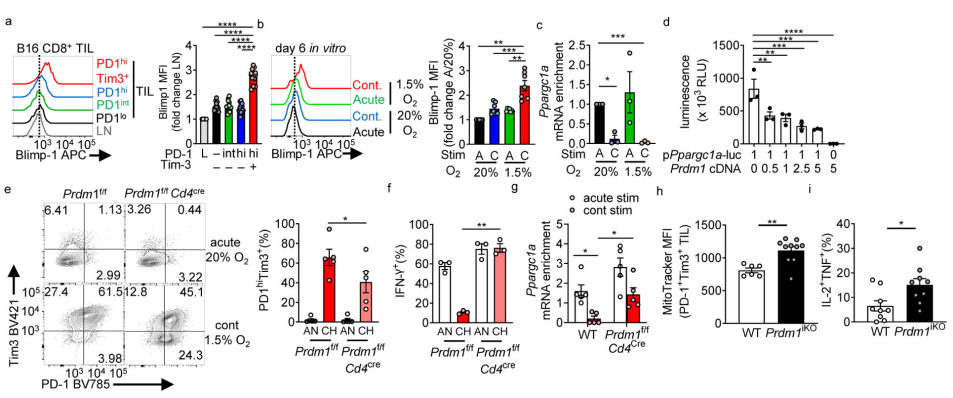
持久抗原对于改变向衰竭分化至关重要,但其代谢后果尚不清楚。由于连续的刺激不产生明显的代谢抑制,持续的刺激可能激活一个转录抑制因子,阻止代谢重编程。Blimp-1(由Prdm1编码)就是这样一种抑制因子,在B16黑色素瘤中最终耗尽的CD8+ T细胞中高度上调(图4a),并在缺氧条件下持续激活(图4b)。体外缺氧条件下持续激活的T细胞也抑制PGC1α的表达(图4c)。 进一步确定Blimp-1是否可以抑制PGC1α,发现强制表达Blimp-1可以抑制293T细胞中Ppargc1a启动子构建的活性,而不影响其活力(图4d)。Prdm1f/fCd4Cre小鼠的CD8+ T细胞通过在缺氧条件下持续激活而抵抗功能障碍(图4e,f),而在持续激活条件下无法抑制Ppargc1a的表达(图4g)。在PD-1hiTim3+ TILs中,Blimp-1的缺失导致了通过IL-2和肿瘤坏死因子(TNF)产生的线粒体质量和多功能性的恢复(图4h, i)。
五、ROS通过作为磷酸酶抑制剂来驱动衰竭
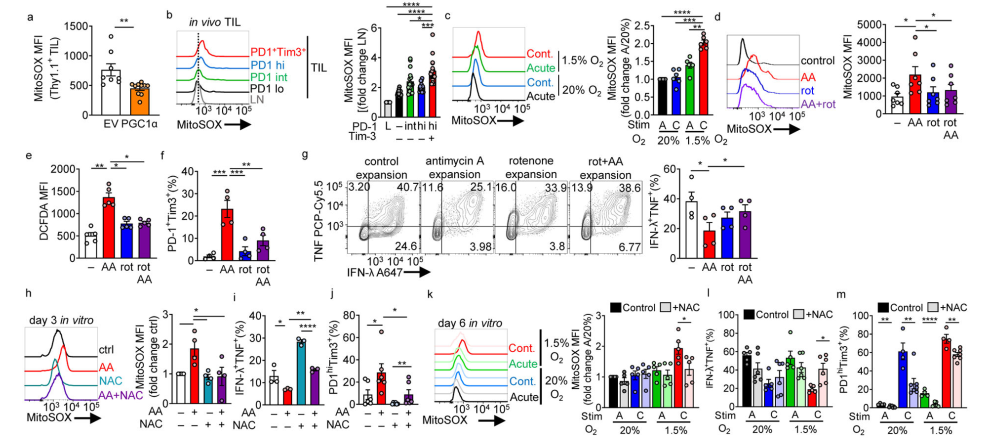
肿瘤浸润PGC1α oe Pmel-1 T细胞在肿瘤浸润时线粒体ROS减少(图5a),表明PGC1α部分作用于肿瘤浸润时ROS的减少。 内源性B16 TIL检查显示,最终耗尽的T细胞含有大量的mtROS(图5b)。体外缺氧条件下的持续激活也产生了高水平的mtROS(图5c),表明ROS可能是T细胞衰竭的驱动因素(图5d,e)。低、无毒剂量的药物用于培养T细胞数天,激活T细胞(24小时),然后在抗霉素A存在的情况下扩增,会导致衰竭样功能障碍:共同抑制分子高表达,多功能性减少(干扰素-g (IFN-γ)和TNF的产生)(图)。5 f, g)。添加鱼藤酮,当添加到抗霉素A处理时,整个电子传递链崩溃,挽救了功能障碍,表明所观察到的衰竭不是由于线粒体功能的丧失,而是由于线粒体应激和随后的ROS(图5d-g)。为了进一步解决ROS驱动功能障碍的作用,我们使用n -乙酰半胱氨酸(NAC),一种中和ROS的细胞渗透性抗氧化剂(图5h)。NAC能够防止抗霉素A或缺氧下持续刺激引起的功能障碍(图5i m)。
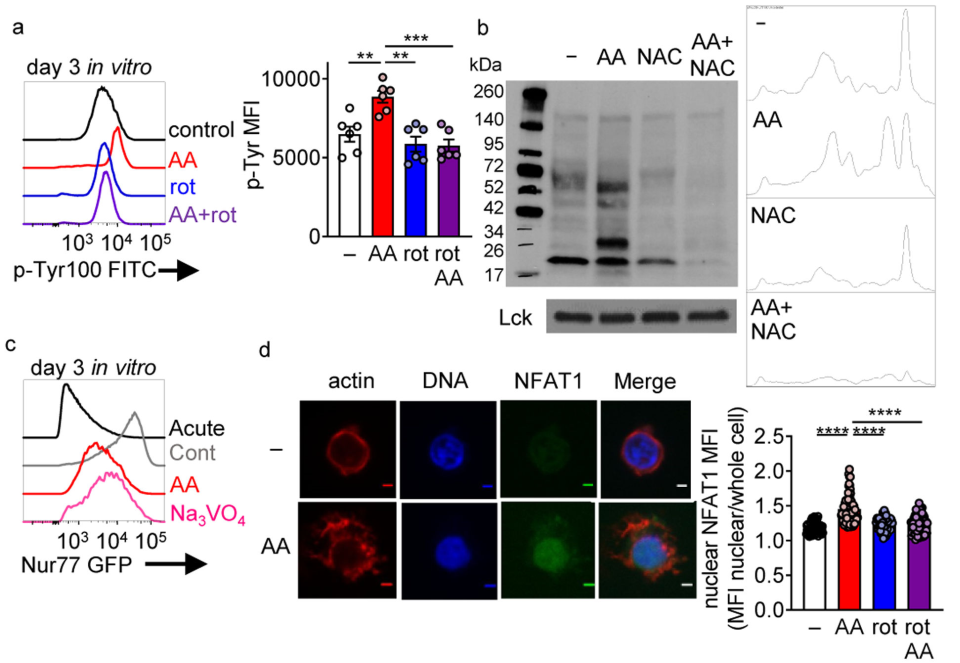
ROS及其细胞副产物是酪氨酸磷酸酶的有效抑制剂,、用抗霉素A培养T细胞可导致酪氨酸磷酸化的持续和升高(图6a),这被鱼藤酮或NAC阻断(图6a,b)。免疫印迹分析显示,在aa诱导的ROS下,酪氨酸磷酸化的绝对数量增加,但几种不同的蛋白在NAC处理下有差异的磷酸化(图6b)。TCR-reporter Nur77-GFP小鼠研究表明在TCR信号缺失的情况下,抗霉素A诱导GFP表达;在低于连续刺激条件下诱导的信号时,抗霉素A处理产生的GFP与酪氨酸磷酸酶抑制剂orthovandate (50 μm)培养的T细胞表型相匹配(图6c)。磷酸酪氨酸级联促进NFAT1的核积累,单独的ROS诱导(通过抗霉素A)以鱼藤酮抑制的方式促进NFAT1核的增加(图d).
六、减轻ROS或缺氧可缓解T细胞衰竭

Pmel-1 T细胞被转导Gpx1, Gpx1是一种谷胱甘肽过氧化物酶和已知的能作用于许多ROS物种的PGC1α靶点,然后转移到携带b16的动物体内。与pgc1 α转导的细胞一样,gpx1过表达的T细胞对肿瘤中ROS的积累具有抗性(图7a)。过表达gpx1的TIL T细胞保持功能,产生更多的ifn - γ(图7b)。因此,通过细胞固有的方式减少ROS可以保护T细胞免受肿瘤诱导的衰竭。Ndufs4缺乏的肿瘤在体外没有明显的氧消耗,在体内产生较少的缺氧(图7c,d)。在这些内源性的浸润性CD8+ T细胞中,较小比例的细胞发展到最终衰竭(图7e)。然而,虽然这些细胞仍然表达多种共抑制分子,但浸润Ndufs4缺陷肿瘤的PD-1hiTim3+ T细胞显示多功能性增加(图7f)。用低剂量(10 mg/kg)阿西替尼治疗b16小鼠降低了肿瘤总量和用吡莫唑测量的T细胞缺氧(图7g,h)。瘤内T细胞表型上耗竭较少(图7i),多功能性较多(图7j),提示通过靶向VEGFR和降低缺氧,T细胞可能对免疫治疗更敏感。在体内用低剂量阿西替尼治疗荷瘤小鼠可使荷瘤小鼠对CTLA-4和PD-1阻断剂致敏,降低肿瘤负担并提高生存率(图7k)。通过靶向肿瘤微环境的缺氧特性,T细胞不会分化到终末衰竭,并保持对检查点封锁的反应。