






铁死亡诱导Ca2+通量和ESCRT-III激活来调节细胞死亡动力学

铁死亡(Ferroptosis)是一种不依赖caspase的调控细胞坏死形式,其特征是在细胞膜上产生铁依赖的脂质过氧化物。Ferroptosis发生的一个基本步骤是破坏质膜。然而,导致Ferroptosis质膜完整性丧失的分子机制以及膜损伤的性质和大小尚未得到深入研究。其他形式的细胞死亡如坏死、焦亡或毒素诱导的细胞死亡会导致离子通量的激活。在这些情况下,Ca2+通量与膜修复机制的激活有关,同时转运体(ESCRT)发挥了关键的平衡作用,可以延缓坏死和焦亡症中的细胞死亡。目前有作者研究了不同细胞模型中Erastin-1和RSL3引发的Ferroptosis中质膜渗透的分子机制。利用活细胞成像和流式细胞术,跟踪了Ferroptosis不同特征的动力学。该研究发表于2021年3月《Cell Death & Differentiation》期刊上,IF:10.717。
技术路线:

主要结果如下:
1. 细胞内Ca2+持续增加是ferroptosis的标志
胞浆Ca2+增加、细胞肿胀以及质膜破裂被认为是坏死和焦亡的标志。为了评估这些细胞改变是否也发生在ferroptosis期间,作者通过活细胞共聚焦成像(图1A-B)和流式细胞术(图1C-H)检测了用Erastin-1或RSL3处理的小鼠成纤维细胞(NIH-3T3)的胞浆Ca2+水平,同时检测了细胞形态和质膜破裂的变化。作者使用fluo-4am作为Ca2+指示剂的荧光形式,以可视化细胞内Ca2+水平,并使用荧光DNA插入剂PI作为不可逆质膜破坏和细胞死亡的标记。结果发现在使用Erastin-1或RSL3处理后,NIH-3T3细胞胞质Ca2+浓度明显增加(图1A, B)。胞浆内Ca2+增加、细胞圆化和质膜破裂可被ferroptosis特异性抑制剂ferrostatin-1(fer1)抑制(图1A-F)。
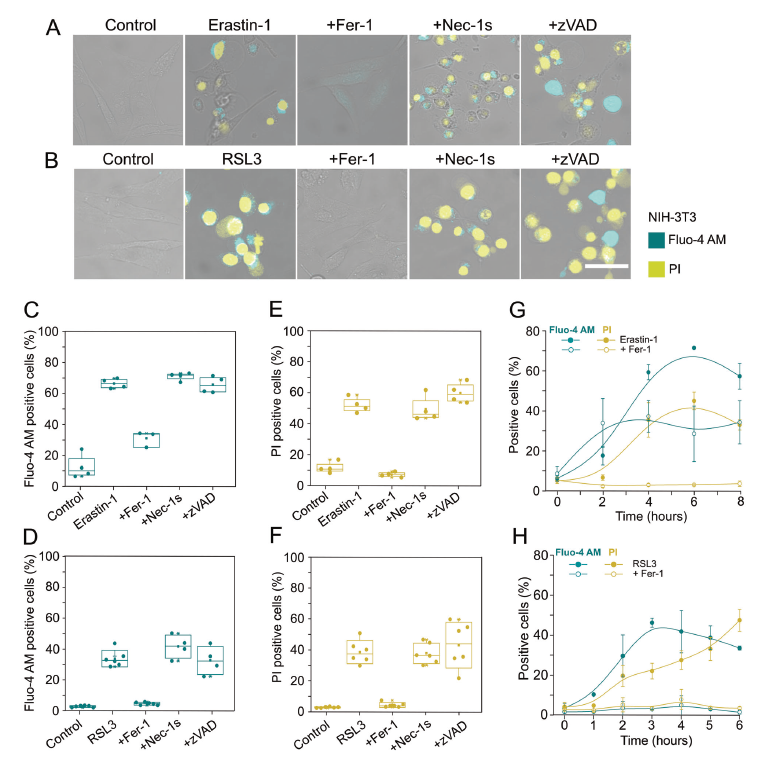
图1 Ferroptosis伴随着细胞内Ca2+增加和细胞死亡
2. 脂质氧化先于胞质Ca2+的增加和质膜的破坏
体积细胞实验中Ferroptosis特征之间的详细时间关系被治疗后群体细胞发生Ferroptosis的异质性所掩盖。为了解决这个问题,作者使用活细胞共聚焦显微镜在单细胞水平上量化了Fluo-4 AM信号、细胞圆度和PI摄入量的增加。从单个细胞获得的动力学曲线(图2C, F, K)中,计算出每个ferroptotic表型(t50)实现50%变化所需的时间,这使得可以设置Ferroptosis各过程之间的滞后时间(图2D, G, L)。结果发现,与所使用的Ferroptosis触发器无关,胞浆Ca2+的增加先于细胞圆缩和质膜完全坍塌(图2A, B)。从流式细胞术实验(图1G)可以看出,Erastin-1存在时,胞质Ca2+的增加是一个双相行为的两步过程(图2C)。第一个事件在最大Ca2+信号的40%左右达到饱和,与不相关的钙(Ca2+un)增加相对应,因为它没有受到Ferroptosis的抑制(图1G)。第二次Ca2+升高,与RSL3处理常见,并被Fer-1抑制,对应的是针对铁下垂的胞质Ca2+增加(Ca2+sp) (图2F)。在Erastin-1处理后,Ca2+un增加,随后Ca2+sp上升,细胞周围和最终的PI摄入(图2C-E)。相比之下,RSL3处理的细胞具有独特和特定的Ca2+上升,随后细胞圆整和最终的PI摄入(图2F-H)。通过脂质过氧化传感器C11 BODIPY 581/591观察到RSL3处理促进了质膜和亚细胞膜上的脂质过氧化(图2I),脂质过氧化先于细胞圆化(图2J, K)。考虑到胞质Ca2+增加到细胞圆化之间的滞后时间始终低于脂质过氧化到细胞圆化之间的滞后时间,可以估计Ca2+的增加发生在脂质过氧化之后(图2M)。这一估计是基于fluo- 4am(图2F-H)和BODIPY氧化比(图2K, L)的独立动力学之间的推断。
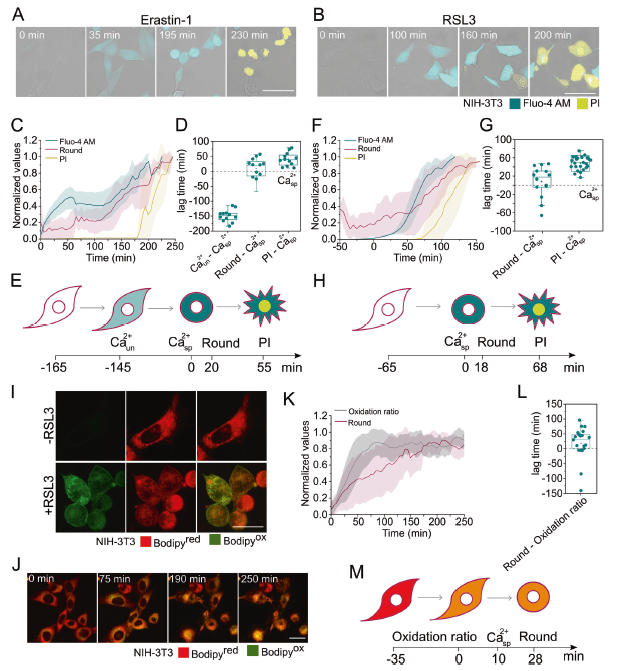
图2 在质膜破裂之前,胞质Ca2+和脂质氧化增加
3. 渗透活性剂保护细胞防止ferroptosis
气孔形成是不同类型调控细胞死亡的共同特征。质膜孔的打开导致水分子的流入,这是由于无法通过膜孔的高浓度细胞内大分子造成的渗透失衡的结果(图3A)。这种效果可以通过添加不能够通过孔进入细胞的适当大小的渗透保护剂peg来阻止,从而平衡细胞内渗透压、水流入和随后的细胞坍塌(图3B)。加入1.8 nm的peg并不能阻止胞质Ca2+的增加、细胞圆化或细胞死亡(图3C-F)。相比之下,在peg(2.3nm和2.7nm)的存在下,胞质Ca2+水平的增加和细胞死亡均以尺寸依赖的方式延迟(图3D, F)。PEG(2.7nm)对ferroptosis的保护作用在洗涤后恢复,这表明膜损伤随时间的推移是稳定的(图3G)。我们还发现,PEG(2.7nm)并不能阻止RSL3处理的NIH-3T3细胞的脂质过氧化(图3H, I)。对于使用的所有PEG大小,NIH-3T3细胞处理较长时间(24 h)后细胞死亡恢复(图3J)。PEG对ferroptosis动力学的抑制作用也随着RSL3浓度的增加而降低(图3K, L)。最终作者以在较低浓度RSL3下能够起到保护作用的PEG尺寸作为参考,得出质膜穿孔部分的半径约为2.5 nm(图3L)。
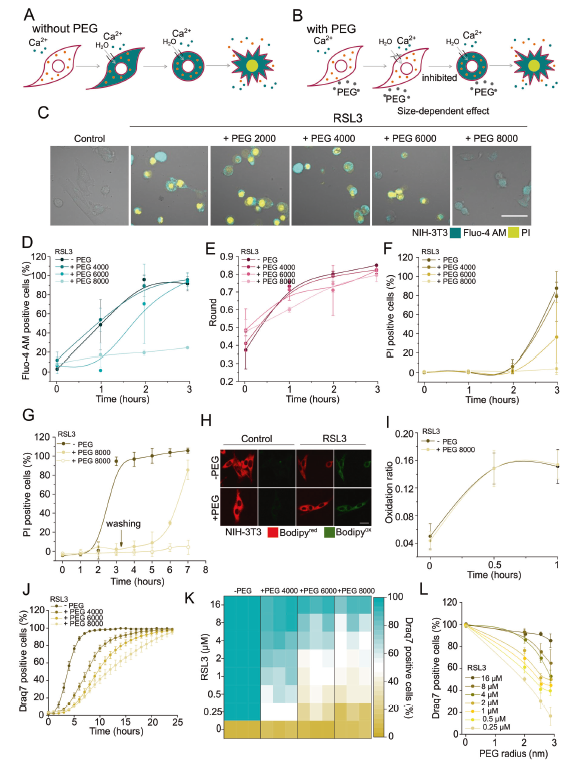
图3 大尺寸的PEG对ferroptosis有渗透保护作用
4. ESCRT-III复合物在ferroptosis期间被激活,并拮抗细胞死亡
在坏死和焦亡过程中,依赖于ESCRT-III复合物作用的膜修复可以延缓细胞死亡。作者使用CHMP4B斑点形成作为代替ESCRT-III激活。最初,CHMP4B主要表现为均匀的胞质分布,然而,在RSL3处理后,该蛋白定位于不同的点状结构,随着时间的推移,其数量和荧光强度都在增加(图4A, B)。CHMP4B点的形成大致与胞质Ca2+浓度的增加相一致(图4C, E)。当细胞修复机制最终被淹没时,细胞内Ca2+水平持续升高、细胞圆化和ESCRT-III复合体激活后,细胞膜最终破裂(图4B, E, F)。PEG(2.7nm)处理也阻断了CHMP4B斑点的形成 (图4G-J)。这些结果证明了ESCRT-III复合物以Ca2+依赖的方式被激活来修复膜损伤。
为了研究ESCRT-III在ferroptosis中的功能作用,作者评估了抑制CHMP4B对NIH-3T3细胞死亡的影响(图5A, B)。与对照组相比,CHMP4B敲除细胞的死亡发生率更高、更快,特别是在早期时间点(1-3 h),这表明CHMP4B的缺失导致细胞对ferroptosis的敏感性更高。另外,作者使用蛋白质组谱仪XL细胞因子阵列试剂盒(图5C),确定ferroptosis是否可以直接调节不同细胞系和不同处理下的细胞因子分泌。在诱导ferroptosis时,作者检测到细胞因子亚群水平的变化,这些变化与细胞系和治疗有关。此外,敲除CHMP4B改变了RSL3处理后获得的细胞因子谱(图5C, D)。这些结果表明,尽管具体的细胞因子改变依赖于治疗,但ESCRT-III通常影响ferroptotic细胞的细胞因子分泌。另外,RSL3处理的沉默CHMP4B细胞的上清能够增加小鼠巨噬细胞的CD14表面表达(图5E)。这些结果表明,与坏死和焦亡相似,ESCRT-III也调节ferroptotic细胞的免疫输出。

图4 膜损伤与CHMP4B活化有关
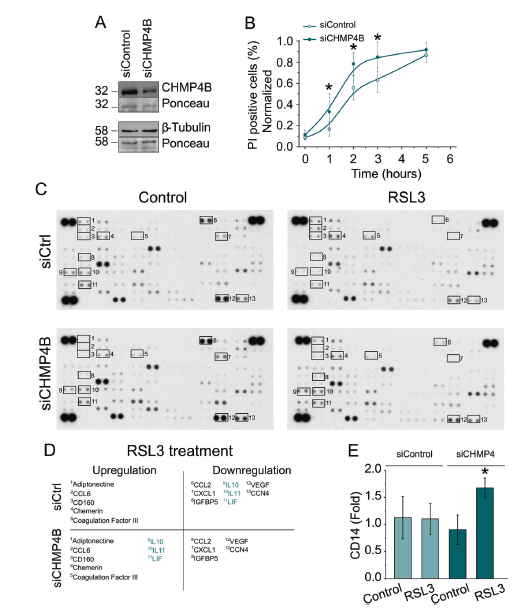
图5 ESCRT-III调节细胞死亡和嗜铁细胞的免疫结果
结论:
ESCRT-III在ferroptosis微环境中调节细胞因子水平,揭示了这一机制在调控ferroptosis炎症的作用。这些发现支持了一个普遍的模型,在这个模型中,质膜孔的形成和膜修复是控制不同类型的坏死及其炎症特征的中心和相反的调控机制。
参考文献:
Pedrera L, Espiritu RA, Ros U, Weber J, Schmitt A, Stroh J, Hailfinger S, von Karstedt S, García-Sáez AJ. Ferroptotic pores induce Ca2+ fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021, 28(5):1644-1657. doi: 10.1038/s41418-020-00691-x