






SIRT1与CRL4B复合物协同调控胰腺癌干细胞特性促进肿瘤发生
胰腺癌是一种常见的恶性肿瘤,预后较差。最近,肿瘤干细胞(CSCs)被发现存在于包括胰腺癌在内的几种实体肿瘤中。尽管越来越多的证据表明sirtuin 1 (SIRT1)在各种癌症中发挥着生物学功能,但它是如何参与胰腺癌的发生和转移,以及它在CSCs中的作用仍不明确。2021年6月发表于“Cell Death & Differentiation”(IF=15.828)的文章“SIRT1 coordinates with the CRL4B complex to regulate pancreatic cancer stem cells to promote tumorigenesis”对此展开了研究。研究发现SIRT1与CRL4B复合物相互作用,该复合物负责H2AK119泛素化。SIRT1/CUL4B靶点的全基因组分析确定了一组基因,包括GRHL3和FOXO3,它们在细胞分化、生长和迁移中起关键作用。此外,我们发现SIRT1和CUL4B共同促进胰腺癌细胞的增殖、自噬和侵袭。值得注意的是,我们发现SIRT1/CUL4B促进CSC样特性。体内实验表明,SIRT1促进了已建立的肿瘤异种移植瘤的生长,增加了NOD/SCID小鼠的肿瘤起始能力,增加了CSC频率。值得注意的是,SIRT1和CUL4B的表达在包括胰腺癌在内的多种人类癌症中显著上调。

技术路线
结果:
1)sirtuin对胰腺癌细胞干细胞样表型影响的系统分析
7个人类sirtuin成员(SIRT1-7)共享一个保守的NAD+依赖的去乙酰化酶结构域(图1A)。为了研究sirtuins是否影响胰腺癌细胞的干细胞样表型,flag标记的SIRT1-7在PANC-1细胞中稳定表达。过表达SIRT1的细胞中CD133+细胞含量增加(图1B),其他细胞无明显变化。过表达SIRT1的PANC-1和AsPC-1细胞中干细胞标记物上调,而过表达其他sirtuin家族成员只改变了部分CSC标记物的表达(图1C和D)。SIRT1敲低后,这些因素的表达下降。此外,敲低SIRT2-7并没有引起这些CSC标记物表达的统一变化(图1E和F)。综上所述,这些结果表明SIRT1似乎与CSC相关的特性有关,如CD133表达和干性基因水平。

2)SIRT1与CRL4B复合体相关
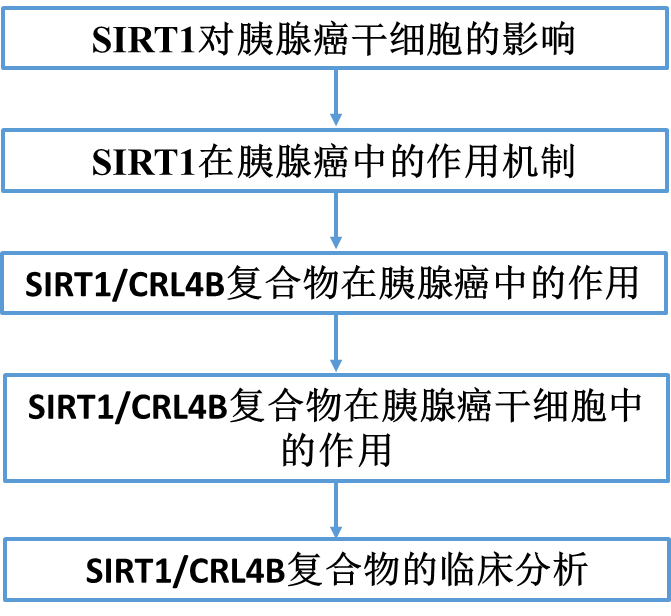
为了更好地理解SIRT1在胰腺癌中的作用机制,我们使用亲和纯化和质谱分析,结果显示SIRT1与各种表观遗传因子共纯化(图2A)。在列出的蛋白中,SIRT1与MTA1、HDAC1/2、EED和SAP30的关联已有报道。western blot进一步证实了SIRT1相关复合物中存在这些蛋白(图2B)。除了先前报道的与SIRT1相互作用的蛋白外,新发现的SIRT1相关蛋白DDB1表明,SIRT1可能与CRL4B复合物的组成部分CUL4B和ROC1在物理上相互作用(图2B)。为了进一步证实SIRT1和CRL4B复合物在体外的相互作用,我们在四种胰腺癌细胞株中进行了co-IP检测,结果显示SIRT1与CRL4B复合物共免疫沉淀(图2C)。为了确定SIRT1/CRL4B复合物的存在,我们使用FPLC对核蛋白进行了蛋白分离实验。Western blotting显示,SIRT1的洗脱模式与CRL4B组分的洗脱模式大部分重叠(图2D),支持了SIRT1和CRL4B复合物可能在体内有功能合作的论点。接下来,GST下拉分析结果显示SIRT1直接与CUL4B和DDB1相互作用(图2E)。此外,SIRT1的N-terminal负责CUL4B的结合,而C-terminal是DDB1的结合所必需的(图2F(a))。结果还表明,CUL4B NEDD8结构域直接参与与SIRT1相互作用(图2F(b))。CUL4B类泛素化位点先前已报道在氨基酸836-843上。结果显示,具有泛素化位点缺失的CUL4B NEDD8结构域仍然直接与SIRT1相互作用(图2F(c)),表明SIRT1与CUL4B的相互作用不依赖于类泛素化修饰。GST下拉分析表明,DDB1 BPC结构域与SIRT1相互作用(图2F(d))。这些结果不仅进一步支持了SIRT1与CRL4B复合物之间的特异性相互作用,而且揭示了SIRT1/CRL4B复合物形成的分子机制(图2G)。
为了进一步探索SIRT1和CUL4B之间的功能关系,我们研究了SIRT1或CUL4B是否会改变H3K9ac、H3K14ac、H4K16ac和H2AK119ub1的整体水平。Western blot结果显示,过表达SIRT1降低了H3K9ac、H3K14ac、H4K16ac,而显著增加了H2AK119ub1(图2H)。在PAN1细胞中敲除SIRT1,这些组蛋白位点表现出相反的趋势。CUL4B过表达显著增加H2AK119ub1,降低H4K16ac;CUL4B下调后,H2AK119ub1显著降低,H3K9ac、H3K14ac和H4K16ac略有升高(图2H)。这些结果提供了支持SIRT1和CRL4B复合物相互作用的特定机制的证据;从而确定SIRT1和CUL4B的功能连通性。

3)SIRT1/CRL4B复合物转录靶点的全基因组识别
接下来,我们使用ChIP-seq分析全基因组SIRT1/CRL4B复合物转录靶点。我们发现SIRT1和CUL4B特异性结合峰分别为10455和12591(图3A)。此外,我们发现SIRT1和CUL4B具有相似的结合基元(图3B),这支持了它们在物理上相互作用并在功能上相互连接的观点。然后,对SIRT1(1560个基因)和CUL4B(2369个基因)的DNA启动子序列进行重叠分析;这些启动子代表了SIRT1/CRL4B复合物的共同靶点(图3C)。我们鉴定了288个SIRT1和CUL4B特异性启动子,并将它们分为不同的细胞信号通路,包括Rap1、AMPK、FoxO、Hippo、细胞周期、黏附、以及调控干细胞的多能性,这在肿瘤的形成和发展中起着至关重要的作用(图3D)。qChIP分析显示,SIRT1和CUL4B对FOXO3、GRHL3、NAV3、AF6、PRDM2、MOB1A、DLG1、CTNNA1和CTNNA3等经典通路基因的启动子具有强富集作用(图3E)。RT-qPCR进一步显示,SIRT1或CUL4B敲低后,PANC-1细胞中靶基因的转录水平部分升高(图3F)。ChIP实验表明,SIRT1或CUL4B缺失的PANC-1细胞中,SIRT1和CUL4B向其靶启动子的募集均减少(图3G和H)。因此,SIRT1和CRL4B可能相互促进对方在靶启动子上的募集或稳定,形成抑制靶基因表达的转录抑制复合物。

4)通过SIRT1/CRL4B复合物调控FOXO3和GRHL3
我们评估了靶向SIRT1和CUL4B mRNA的慢病毒shRNA(图4A),选择最有效的进行接下来的实验。FOXO3是一个成熟的肿瘤抑制基因,参与多种细胞过程;GRHL3是分化所必需的,并具有抑制肿瘤的作用。因此,我们研究了SIRT1/CRL4B复合物对FOXO3和GRHL3的转录调控。SIRT1或CUL4B敲低导致PANC-1(图4B和C)和AsPC-1(图4D和E)细胞中FOXO3和GRHL3的表达增加。在PANC-1细胞中,通过ChIP或ChIP/Re-ChIP实验进一步研究了SIRT1/CRL4B复合物介导的FOXO3和GRHL3的调控(图4F)。这些结果支持了SIRT1和CRL4B复合物作为一个蛋白复合物占据FOXO3和GRHL3启动子。此外,qChIP分析表明,敲除SIRT1、CUL4B或DDB1的表达导致FOXO3和GRHL3启动子相应蛋白的募集显著减少(图4 G)。SIRT1下调不仅导致FOXO3和GRHL3启动子H3K9ac、H3K14ac、H4K16ac升高,还显著降低了H2AK119ub1水平。CUL4B或DDB1基因敲低也得到类似的结果,表明SIRT1/CRL4B复合物作为一个整体与FOXO3和GRHL3启动子结合,催化组蛋白的泛素化和去乙酰化(图4G)。这进一步证实了SIRT1和CRL4B复合物通过一组靶基因(如FOXO3和GRHL3)的转录抑制在功能上存在关联。
5)SIRT1和CUL4B共同促进胰腺癌细胞的增殖、自噬和转移
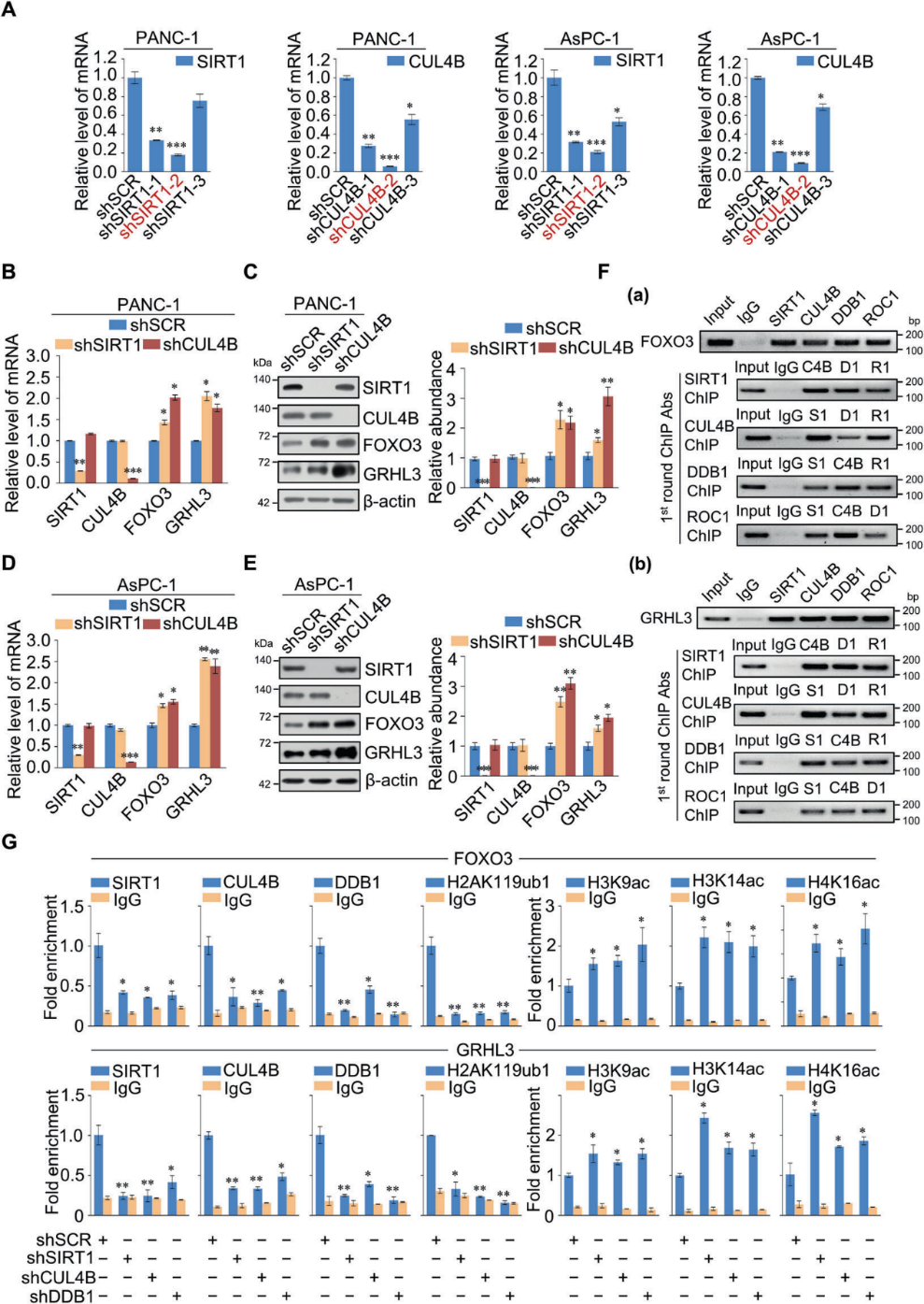
我们研究了SIRT1/CRL4B复合物在胰腺癌细胞增殖、自噬和转移中的作用。生长曲线分析显示,SIRT1和CUL4B促进细胞增殖(图5A)。EdU结果显示,在EdU标记的细胞中,SIRT1或CUL4B过表达与明显的百分比增加相关,而SIRT1或CUL4B敲低的细胞显示这些细胞的百分比要低得多(图5B)。这表明SIRT1/CRL4B复合物在体外促进了胰腺细胞的增殖。
有报道称自噬阻断降低了胰腺CSC活性。因此,我们接下来研究了SIRT1和CUL4B在自噬中的作用。在EGFP-LC3 PANC-1细胞中,SIRT1或CUL4B过表达增加LC3斑点(图5C)。在mCherryGFP-LC3 PANC-1细胞中,SIRT1或CUL4B过表达显示GFP-/mCherry+(红色斑点)自噬溶酶体增加,在较小程度上,GFP+/mCherry+(黄色斑点)吞噬体/自溶噬体增加(图5D)。接下来,用LysoTracker Red对溶酶体隔室进行染色,显示过表达SIRT1或CUL4B的PANC-1细胞中溶酶体区域扩大(图5E)。此外,流式细胞术显示过表达SIRT1或CUL4B的细胞中LysoTracker+细胞含量增加(图5F)。我们使用自噬体和溶酶体融合抑制剂巴菲霉素A1 (BafA1)进行自噬通量分析。经过BafA1处理后,SIRT1和CUL4B的增加与LC3-II的积累相关。western blot分析也证实了SIRT1和CUL4B基因下调导致的自噬通量缺陷(图5G和图S3C)。这些结果表明,SIRT1或CUL4B过表达不仅增强了自噬通量,还增强了溶酶体功能。
接下来,western blot显示上皮标记物,如α-和γ-catenin表达减少,而间质标记物,包括N-cadherin和Vimentin,在SIRT1或CUL4B过表达时增加(图5H)。在PANC-1和AsPC-1细胞中单个敲除SIRT1或CUL4B时,这些EMT标记物表现出相反的趋势。此外,在PANC-1和AsPC-1细胞中进行transwell侵袭检测结果显示,过表达SIRT1或CUL4B导致细胞侵袭能力增加超过两倍,而敲除SIRT1或CUL4B则导致细胞侵袭能力明显降低(图5I)。下调CUL4B或SIRT1后,过表达SIRT1或CUL4B的作用减弱,同时下调SIRT1和CUL4B后,细胞侵袭能力明显减弱(图5I)。在PANC-1细胞中敲低SIRT1或CUL4B降低了细胞侵袭潜能,这部分可通过共同敲低FOXO3或GRHL3来挽救,表明SIRT1/CRL4B复合物可通过抑制FOXO3和GRHL3来促进胰腺癌侵袭(图5J)。这些结果表明,SIRT1/CRL4B复合物促进胰腺癌细胞的迁移和侵袭潜能,部分通过抑制FOXO3和GRHL3。
6)SIRT1和CUL4B促进胰腺癌干细胞特性
我们研究了SIRT1和CUL4B是否影响胰腺癌细胞的干细胞样表型。在稳定表达SIRT1或CUL4B的PANC-1和AsPC-1细胞中,干细胞标记物均增加(图6A)。为了进一步阐明SIRT1和CUL4B是否促进胰腺癌细胞向CSCs的发展,我们分析了SIRT1和CUL4B对球体形成的影响。在稳定表达SIRT1或CUL4B的PANC-1和AsPC-1细胞中,球的数量和大小增加,而下调SIRT1或CUL4B后,球的数量和大小减少(图6B)。接下来,流式细胞术显示过表达SIRT1和CUL4B后CD133+细胞数量增加,这一效应在抑制CUL4B和SIRT1后部分被挽救(图6C)。这些结果表明SIRT1/CRL4B复合物促进胰腺CSC特性。为了进一步证实SIRT1在体内靶向胰腺CSC群体,我们建立了小鼠异种移植模型。注射4周后,通过生物发光观察移植细胞的生长情况(图6D)。稳定表达SIRT1的细胞显著增加了肿瘤起始能力(图6E)。SIRT1过表达组与对照组相比CSC频率显著增加(图6F)。此外,SIRT1显著促进胰腺肿瘤的生长(图6G)。这表明SIRT1可以显著诱导胰腺癌细胞的干细胞性,从而促进肿瘤移植瘤的生长。与肿瘤生长加速一致,SIRT1过表达组Ki67阳性细胞比例明显高于对照组(图6H)。我们利用RNA-seq研究了SIRT1过表达的全基因组效应。与对照组相比,我们在SIRT1过表达的肿瘤样本中共发现了2506个上调基因和2229个下调基因(图6I)。KEGG富集分析结果显示,这些差异表达的基因不仅参与病灶黏附、Wnt、Hippo、细胞周期等与肿瘤生长和干性密切相关的途径,而且在与自噬相关的吞噬小体和溶酶体途径中富集(图6J)。接下来,我们选择了12个已知的肿瘤抑制因子与癌症发展有关的基因,包括GRHL3、NAV3、AXIN2、FOXP1、WISP3、SPDEF、RASSF1、PTPRG、IGFBP4、FHL1、FEZ1和DUSP4,并通过RT-qPCR验证了它们在SIRT1过表达的肿瘤样本中的表达降低(图6K)。这些结果表明,SIRT1参与调节与肿瘤生长、自噬和干细胞形成密切相关的各种通路,并促进胰腺CSC的发展

7)SIRT1和CUL4B在多种癌症中表达上调,是一种潜在的癌症生物标志物
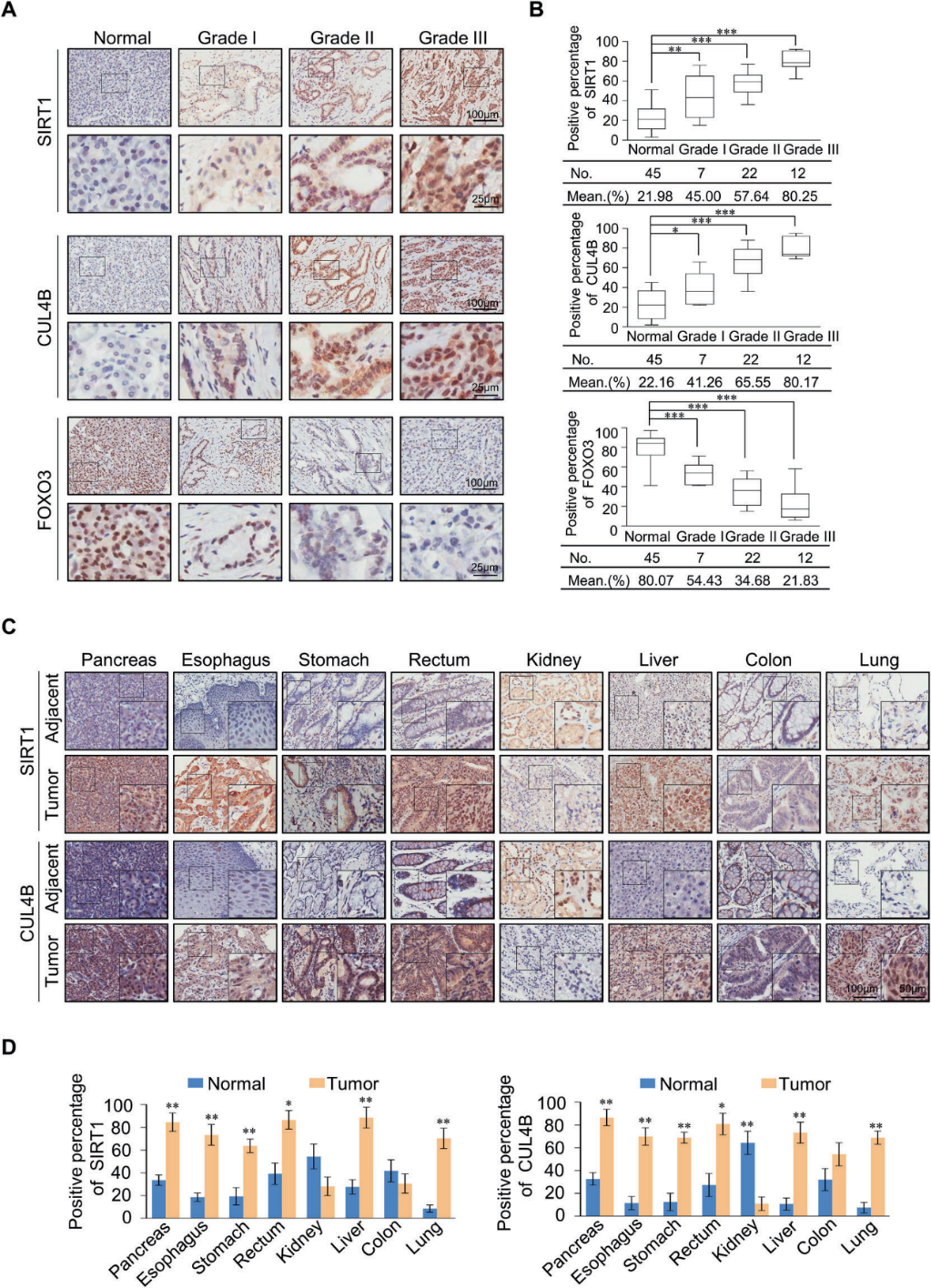
我们收集了86例胰腺癌患者的样本,通过免疫组化染色进行组织微阵列,检测SIRT1、CUL4B和FOXO3的表达情况(图7A)。SIRT1和CUL4B在肿瘤中显著上调,其表达水平与肿瘤组织学分级呈正相关,FOXO3表达水平与肿瘤组织学分级呈负相关(图7B)。为了研究SIRT1和CUL4B的影响是否可以扩展到更广泛的癌症范围,我们收集了几个癌症样本,对其进行组织微阵列和免疫组化染色来检测SIRT1和CUL4B的表达(图7C)。结果显示,除胰腺癌外,SIRT1和CUL4B在食管癌、胃癌、直肠癌、肝癌、肺癌中与癌旁正常组织相比也显著上调(图7D)。总之,我们的分析表明SIRT1和CUL4B在多种癌症中上调,是潜在的癌症生物标志物。
结论: SIRT1和CRL4B作为一个功能单元相互作用和合作,提供了一种新的转录调控模式,同时也为染色质重构中组蛋白去乙酰化和泛素化提供了新的分子基础。此外,SIRT1/CRL4B复合物有助于肿瘤抑制的表观遗传学沉默,也在胰腺癌的肿瘤发生和调节CSCs的特性中发挥重要作用。因此,SIRT1和CUL4B是潜在的致癌基因和生物标志物,可能作为肿瘤治疗的靶点。