






新连接——GOT1抑制通过ferroptosis促进胰腺癌细胞死亡
癌症代谢被重新连接,以支持细胞生存,以应对内在和环境的压力。鉴定靶向这些适应机制的策略是一个活跃的研究领域。作者此前发现GOT1驱动通路在胰腺癌中维持氧化还原平衡。在这里,作者试图鉴定GOT1抑制后的大写依赖以表征胰腺癌的特征并为氧化还原代谢提供新的见解。本文于2021年8月发表在《Nature Communications》IF:14.919期刊上。
技术路线

主要实验结果:
1、PDA细胞利用GOT1促进细胞增殖和肿瘤生长
作者此前的工作证明,胰腺导管腺癌(PDA)细胞重新连接苹果酸-天冬氨酸穿梭线以生成NADPH(Fig. 1a)。为了确定GOT1在PDA中使用时间控制,作者使用dox敲除分别GOT1的编码区和3’UTR(sh1和sh3),以及对照shNT。结果显示sh1和sh3显著敲除GOT1的表达并细胞集落形成(Fig. 1b, c)。在18个细胞系中,有12个细胞系的GOT1敲除显著损害了菌落形成(Fig. 1d)。但是对GOT1敲除的响应不依赖于苹果树-天冬氨酸穿梭酶的表达状态。然后检测GOT1对已建立的小鼠PDA肿瘤的抑制作用。结果显示,dox诱导GOT1敲除后,GOT1敏感细胞株表现出明显的生长抑制(Fig. 1e, f)。这些结果表明PDA利用GOT1促进细胞增殖和肿瘤生长。

图1 对于细胞增殖和肿瘤生长来说GOT1是PDA必须的
2、限制外源性胱氨酸可增强GOT1抑制
由于与非转化细胞相比,GOT1抑制在PDA中具有独特的细胞抑制作用,因此作者试图识别由GOT1敲低诱导的代谢缺陷,从而靶向杀死PDA。为了该目的,作者检测了GOT1敲低PDA细胞对代谢靶向小分子反应的敏感性。对细胞进行5天的dox处理以确保GOT1敲除,然后再进行3天的药物处理(Fig. 2a)。与一些抑制剂联合使用时,GOT1抑制是有保护作用的,表现为曲线值下的面积增加,这是一种药物敏感性的测量(Fig. 2b)。5种最常用的脱敏剂中有3种是化疗药物,这与之前的观察结果一致。相比之下,GOT1敲除增强了erastin的敏感性(Fig. 2b, c)。Erastin是胱氨酸/谷氨酸逆向转运系统的抑制剂,它将胱氨酸转运到细胞中以交换谷氨酸。胱氨酸,半胱氨酸的氧化二聚体,在进入细胞后被还原为半胱氨酸,在细胞内它可以促进谷胱甘肽和蛋白质的合成,以及许多其他生化命运。GOT1敲除增强了PDA细胞系的erastin敏感性(Fig. 2d)并且与dox是否干扰无关。这种GOT1增强效应被erastin类似物咪唑酮erastin (IKE) 复制,而和IKE联合处理可导致部分细胞毒性(Fig. 2e)。与单一处理组相比,erastin或IKE处理GOT1敲除细胞减少了细胞数量,但这可以通过补充外源性半胱氨酸、谷胱甘肽来源GSH-EE或NAC而反转(Fig. 2f)。这与胱氨酸导入到系统xc维持谷胱甘肽水平至关重要的结果一致。在PDA肿瘤中胱氨酸水平是有限的。在肿瘤相关中培养细胞以胱氨酸时间和剂量依赖的方式增强了GOT1的下调(Fig. 2g)。胱氨酸对GOT1敲除小鼠PDA生长的影响与体外实验结果一致。总之,在GOT1抑制后,PDA培养物对外源性胱氨酸戒断敏感。

图2 GOT1抑制之后细胞活力和生长PDA离不开胱氨酸
3、抑制谷胱甘肽的生物合成可增强GOT1基因下调的生长抑制作用
谷胱甘肽(GSH)可以由半胱氨酸重新合成,也可以由氧化的谷胱甘肽(GSSG)经NADPH还原再生(Fig. 3a)。GOT1抑制导致GSSG和NADP+增加,而GSH和NADPH随细胞系而异。因此,作者推测抑制谷胱甘肽的生物合成可能增强GOT1敲除。GSH合成的限速步骤可以被BSO抑制。在24小时的药物处理后,GOT1的抑制有效地增强了对BSO的敏感性,与单药反应温和效应相比,并且BSO暴露72 h会放大这增强作用(Fig. 3b, c)。抑制GOT1可增强BSO对增殖作用(Fig. 3d)。通过补充外源性GSH-EE或NAC可恢复上述对细胞活力的组合效应(Fig. 3e),表明氧化还原失衡。GOT1敲除和BSO暴露对细胞的组合作用在小鼠体内同样适用(Fig. 3f, g)。总之,数据表明,在GOT1敲除的条件下,PDA需要谷胱甘肽的合成来维持生存和生长。
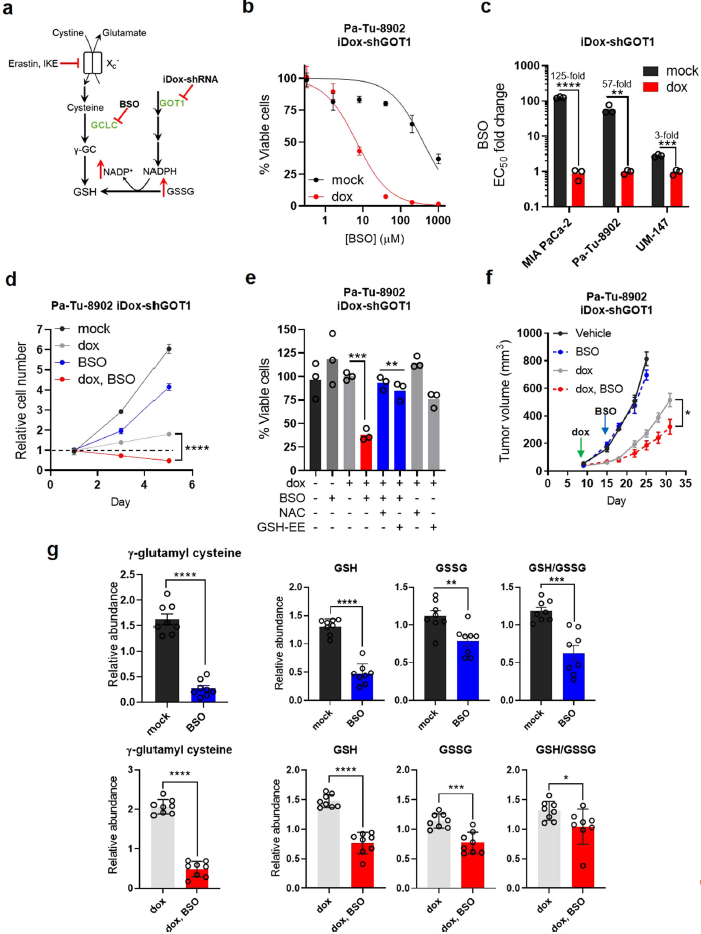
图3在GOT1抑制下,PDA的生长需要谷胱甘肽的合成
4、GOT1抑制增强铁死亡的敏感性
先前的研究表明,某些细胞类型对erastin和BSO敏感,这些药物可以通过消耗GSH来杀死细胞。GSH消耗的近端效应是通过GPX4活性的丧失介导的,GPX4利用GSH作为辅助因子来解毒脂质过氧化物(Fig. 4a)。这可导致脂质过氧化物的致命积累和铁死亡。虽然GOT1抑制不能诱导铁死亡,但本文的数据表明它可能为PDA细胞诱导铁死亡。
为了研究GOT1是否能使PDA对铁死亡敏感,首先检测GOT1与RSL3的组合效应,RSL3是GPX4的共价抑制剂和铁死亡的直接诱导剂。RSL3联合GOT1敲除比单独使用更有效(Fig. 4b)。GOT1增强了一组PDA细胞系的铁死亡,这种作用与dox效应无关(Fig. 4c)。此外,RSL3联合GOT1敲除显著降低细胞增殖而增强了细胞毒性(Fig. 4d)。然后使用C11- BODIPY脂质过氧化传感器来研究GPX4和GOT1抑制如何影响脂质过氧化。单独抑制GOT1可导致适度的脂质ROS,但与RSL3或erastin联合使用可增强脂质ROS的积累;这种增强作用可通过与亲脂抗氧化剂铁抑制素-1 (Fer-1)共同处理逆转(Fig. 4e)。
随后,作者检测了GOT1对铁死亡的作用是否可以通过与缓解脂质过氧化或螯合铁的药物共同处理来抑制。与Fer-1联合处理在可以抑制多个小分子的GOT1增强效应,用亲脂抗氧化剂,Trolox,或铁螯合剂脱铁胺(DFO)处理,可以为细胞提供显著的保护作用(Fig. 4f)。为了排除凋亡、坏死或自噬细胞死亡机制的可能性,用这些细胞死亡途径的特征明确的抑制剂共同抑制GOT1,结果显示,与Trolox或DFO相比,这些抑制剂对细胞的保护作用非常有限。总的来说,这些数据表明,在细胞培养中,GOT1抑制启动了PDA的铁死亡。
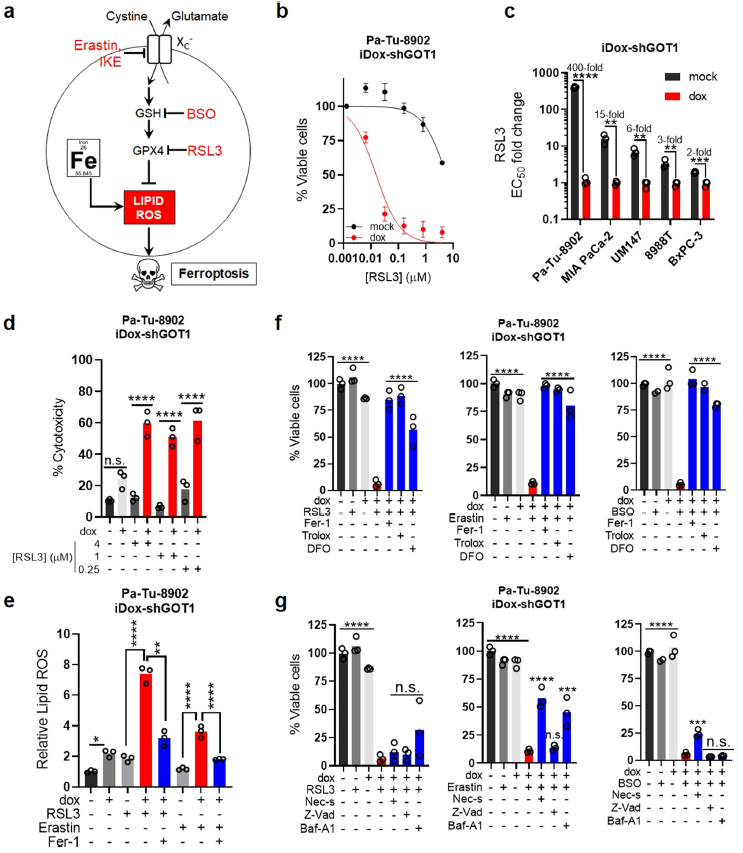
图4 GOT1抑制增强铁死亡
5、GOT1抑制通过促进不稳定铁诱导PDA铁死亡
研究表明代谢紊乱导致细胞内储存的铁自适应释放,以支持线粒体的能量代谢。考虑到铁死亡的易感性与游离铁的可得性有关,作者假设细胞内铁水平可能会随着GOT1抑制介导的能量应激而增加。作者通过测量Calcein-AM来验证这一假设,Calcein-AM是一种荧光素衍生的探针,当与亚铁(Fe2+)结合时被淬灭。GOT1正常细胞的Calcein-AM染色确定基线荧光,GOT1基因敲除后,细胞荧光分布降低,表明活性铁池增加(Fig. 5a)。在培养细胞中使用RhoNox-1铁探针证实了这一观察(Fig. 5b),在皮下和原位肿瘤中观察到总铁水平升高(Fig. 5c)。不稳定铁的影响在表型上很明显,在GOT1缺乏的条件下,需要更高浓度的铁螯合剂DFO来挽救细胞活力(Fig. 5d)。最后,补充柠檬酸铁铵(FAC)可增强铁死亡(Fig. 5e, f)。综上所述,这些数据表明GOT1敲低可促进不稳定铁,铁会增强铁死亡。
GOT1敲低可增强对FINO2(一种药理性铁氧化剂)的敏感性。通过下调铁外流、上调铁摄取或促进细胞内铁载体降解,可以改变铁水平,即铁蛋白(FTN)或血红素。为了检测这些途径中哪一条导致细胞内铁增加,首先检测了铁运输蛋白的表达。GOT1敲低不改变铁运输蛋白SLC40A1的表达。血红素加氧酶1 (HMOX1)的表达在Pa-Tu-8902细胞中未发生改变,但在MIA PaCa-2中表达上调。GOT1敲低对NCOA4和转铁蛋白受体1影响最小。相比之下,IRP2水平较低,FTN水平较高,这两种水平在高铁负荷下均应出现。Pa-Tu-8902细胞的转录组和基因集富集分析都揭示了分解途径的特征通路“溶酶体”和“自噬-溶酶体”。与此一致,GOT1敲除增加了LC3-A/B II/I比值,降低了P62水平,与自噬通量的增加一致(Fig. 5g)。溶酶体抑制剂羟氯喹和Baf-A1说明,GOT1抑制增加自噬通量,而不是延缓自噬过程。
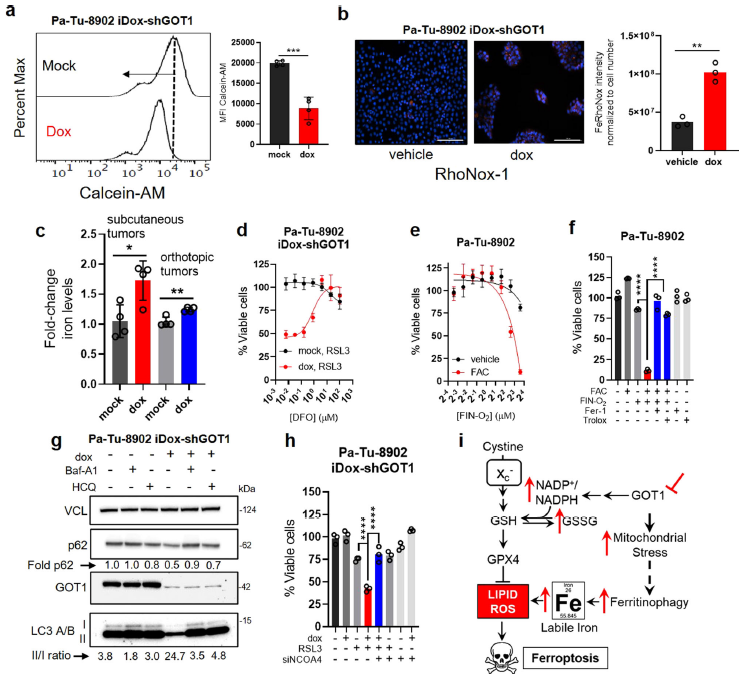
图5 GOT1抑制促进不稳定铁释放
总之,如Fig. 5i所示,抑制GOT1可抑制大量PDA细胞系、原发肿瘤模型和异种移植瘤的生长,同时使一些PDA细胞容易发生铁死亡。抑制胱氨酸输入、GSH合成或GPX4可与GOT1协同触发铁死亡。这种效应可能是由于氧化还原破坏、线粒体抑制和适应性不稳定铁释放的联合作用,所有这些都是已知的增强铁死亡的因素。
参考文献:
Kremer Daniel M., Nelson Barbara S., Lin Lin., Yarosz Emily L., Halbrook Christopher J., Kerk Samuel A., Sajjakulnukit Peter., Myers Amy., Thurston Galloway., Hou Sean W., Carpenter Eileen S., Andren Anthony C., Nwosu Zeribe C., Cusmano Nicholas., Wisner Stephanie., Mbah Nneka E., Shan Mengrou., Das Nupur K., Magnuson Brian., Little Andrew C., Savani Milan R., Ramos Johanna., Gao Tina., Sastra Stephen A., Palermo Carmine F., Badgley Michael A., Zhang Li., Asara John M., McBrayer Samuel K., di Magliano Marina Pasca., Crawford Howard C., Shah Yatrik M., Olive Kenneth P., Lyssiotis Costas A.(2021). GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun, 12(1), 4860. doi:10.1038/s41467-021-24859-2