






ipla2 β介导的脂质解毒控制p53驱动的不依赖GPX4的铁死亡

p53肿瘤抑制因子的失活是大多数癌症形成的关键事件。P53作为一个转录因子,通过调节各种类型的细胞过程来抑制癌症的发展,发挥着核心作用。虽然p53的经典作用包括细胞周期阻滞、凋亡和衰老,这些被认为是肿瘤抑制的主要机制,但最近的研究表明,非传统的机制也对肿瘤抑制有重要作用。最近有研究发现p53通过其代谢靶点在调节癌细胞中ferroptosis反应中发挥重要作用,该研究2021年6月发表在《Nature communications》,IF为12.121。
技术路线:

主要结果:
1. p53诱导的ferroptosis与GPX4无关
为了解决p53介导的ferroptosis是否可以独立于GPX4功能而诱导,作者生成了人骨肉瘤细胞系U2OS的ACSL4/GPX4双敲除衍生物。如图1a所示,ACSL4和GPX4的缺失对p53的水平没有影响,p53介导的p21转录激活或p53介导的SLC7A11抑制仍然完好无损。此外,由于缺乏ACSL4,这些细胞可以抵抗由erastin(图1b)或GPX4抑制剂RSL 3(图1c)诱导的ferroptosis。然而,当ACSL4/GPX4缺失细胞暴露于ROS生成器TBH和p53激活因子Nutlin时,很容易观察到ferroptosis (图1d)。此外,这些p53介导的反应可被已知的ferroptosis抑制剂(如Ferr 1、DFO和Lipro-1)特异性阻断,而不能被其他细胞死亡途径的抑制剂阻断,如凋亡、自噬或坏死(图1d)。
接下来,作者研究了其他ROS生成化合物,如与TBH化学结构非常相似的DDM−9,2-butanone peroxide (BTP)和氢过氧化物异丙苯(CMH),是否可以诱导p53介导的ferroptosis (图2a)。在相同条件下,这两种化合物都容易在亲本和ACSL4/GPX4缺失的U2OS细胞中诱导高水平的ferroptosis (图2b, c)。此外,用C11-BODIPY染色的流式细胞术观察到,当亲本或ACSL4/GPX4缺失细胞分别用Nutlin和TBH处理时,内源性脂质过氧化水平升高,这是ferroptosis的关键标志(图2d, e)。综上所述,这些数据表明p53驱动的铁下垂在高水平ROS下是通过独立于GPX4的方式诱导的。
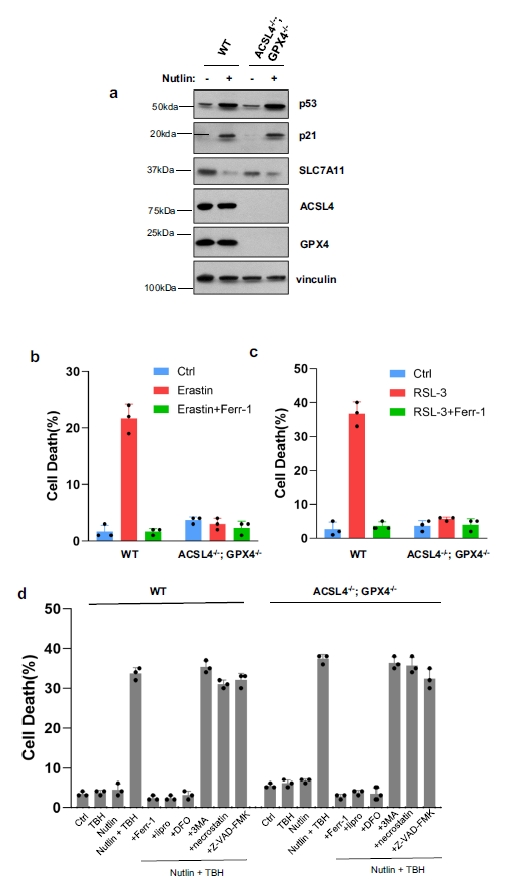
图1 p53介导的ferroptosis不依赖于GPX4诱导的活性氧应激
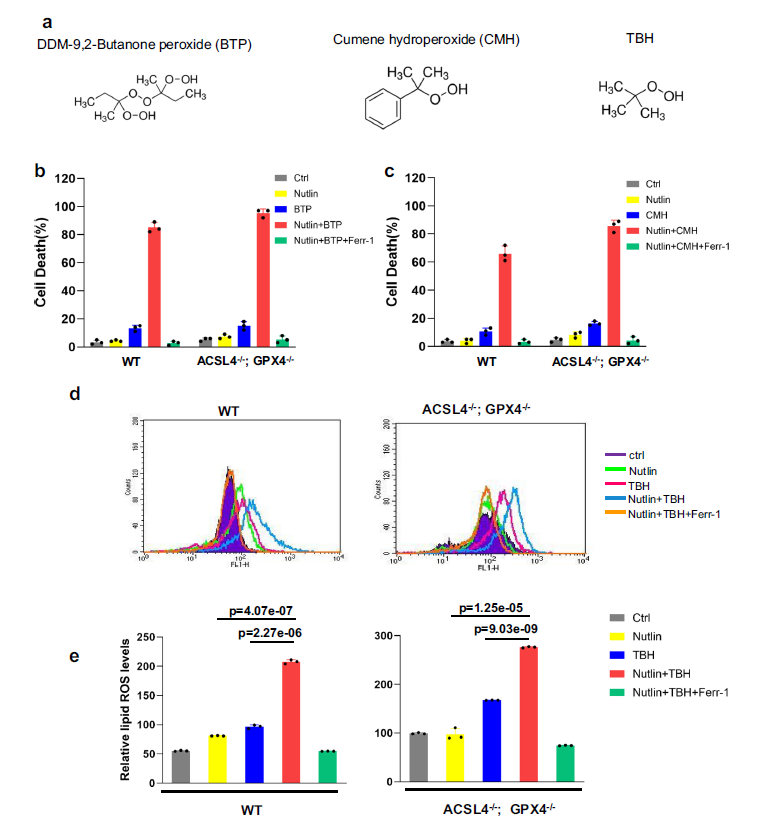
图2 BTP或CMH具有与TBH相似的效应
2. iPLA2β在不同应激水平上受到p53的不同调控
为了确定iPLA2β基因是否确实受p53调控,作者检测了在Nutlin或阿霉素(一种常见的DNA损伤试剂)激活p53时iPLA2β的表达。在表达野生型p53的细胞系(U2OS, MCF7, A375, A549细胞)中,Nutlin或阿霉素处理后iPLA2β mRNA水平轻微但显著上调,但在p53-null细胞系H1299中没有上调(图3a)。如图3b所示,在亲本细胞中,Nutlin处理诱导了iPLA2β的mRNA水平,而不是p53敲除细胞。此外,人类iPLA2β基因的启动子区域包含三个可能的位点(RE1, RE2和RE3),它们与一致的p53结合序列相匹配(图3c)。事实上,p53特异性抗体的染色质免疫沉淀(ChIP)分析显示,p53显著募集到RE2和RE3,而不是RE1,其水平与TIGAR启动子(众所周知的p53代谢靶点)的水平相当(图3d)。值得注意的是,当通过瞬时转染野生型p53重建p53缺失H1299细胞时,iPLA2β mRNA的表达很容易被诱导,但没有被三种肿瘤来源的DNA结合缺陷p53突变体(R175H, R273H,和R248W)诱导(图3e)。
为了进一步了解这一调节的性质,作者检测了在p53介导的应答过程中,相同处理的不同长度或不同剂量下的iPLA2β水平。在Nutlin处理后,iPLA2β水平在较早的时间点被诱导,但激活在较晚的时间点完全消失(图4a, b)。相反,p53介导的p21在所有不同的时间点都被观察到(图4a)。p53对SLC7A11的下调在所有不同的时间点都保持不变(图4a-c)。当细胞同时被Nutlin和TBH处理时,也获得了类似的数据(图4d-f),同时用Nutlin和TBH处理,检测p53介导的ferroptosis。此外,在HCT116细胞中,低剂量的DNA损伤试剂诱导了iPLA2β水平,但高剂量的相同试剂极大程度上消除了iPLA2β的激活,而p53介导的SLC7A11和p21在两种条件下均保持完整(图4g)。

图4 p53介导的iPLA2β的不同效应取决于应激时间和强度
3. 在异种移植小鼠模型中,耗尽内源性iPLA2β可使肿瘤细胞对ROS诱导的ferroptosis敏感,并增强p53依赖的肿瘤生长抑制
为了研究iPLA2β在调节p53功能中的作用,首先检测了RNAi介导的内源性iPLA2β敲低是否会影响人类黑色素瘤A375细胞中p53依赖性ferroptosis。Western blot分析显示,siRNA介导的iPLA2β缺失并不影响p53或其转录靶p21的表达水平(图5a)。转染了对照siRNA的TBH处理的细胞很容易诱导p53介导的ferroptosis (图5b)。然而,在野生型p53中,iPLA2β的消耗显著增加了ferroptosis的水平(图5b),但在相同条件下,在p53阴性细胞中没有观察到显著的影响(图5b),这表明iPLA2β可以抑制p53介导的ferroptosis。如图5c所示,在MCF7细胞中,iPLA2β敲低显著增强了p53介导的ferroptosis。与上述数据一致,iPLA2β表达的缺失对p53水平和p53转录功能没有明显影响(图5d),但在iPLA2β缺失的细胞中,p53介导的ferroptosis在不同时间点显著升高(图5e)。同样,在U2OS细胞中,iPLA2β缺失可显著增强p53介导的ferroptosis(图6f)。
为了阐明iPLA2β在调控铁下垂中的生理意义,检测了iPLA2β表达的缺失是否会影响异种移植瘤模型中的肿瘤生长。在异种移植瘤生长试验中,iPLA2β表达失活后,人类黑色素瘤A375异种移植瘤的生长显著降低(2 vs. 1,图6a, b)。然而,当使用同基因的p53-null A375细胞时,iPLA2β表达缺失诱导的肿瘤抑制作用很大程度上消失了(4 vs. 3,图6a, b)。值得注意的是,在iPLA2β−/−肿瘤样本中观察到Ptgs2的上调,而在iPLA2β−/−/p53−/−样本中没有观察到Ptgs2的上调(图6c)。最后,为了进一步验证这些观察结果,在人肺癌A549细胞中进行了一系列上述类似的实验。同样,iPLA2β的缺失显著增强了p53介导的ferroptosis (图6d)。iPLA2β的失活也促进了p53-WT细胞的肿瘤抑制作用,而不是同基因的p53-null细胞(图6e-g)。综上所述,这些数据表明,抑制iPLA2β表达显著增强了p53介导的ferroptosis,并且通过iPLA2β的缺失激活ferroptosis至少在一定程度上有助于体内p53依赖的肿瘤生长抑制。

图5 iPLA2β作为p53介导的ferroptosis的主要抑制因子
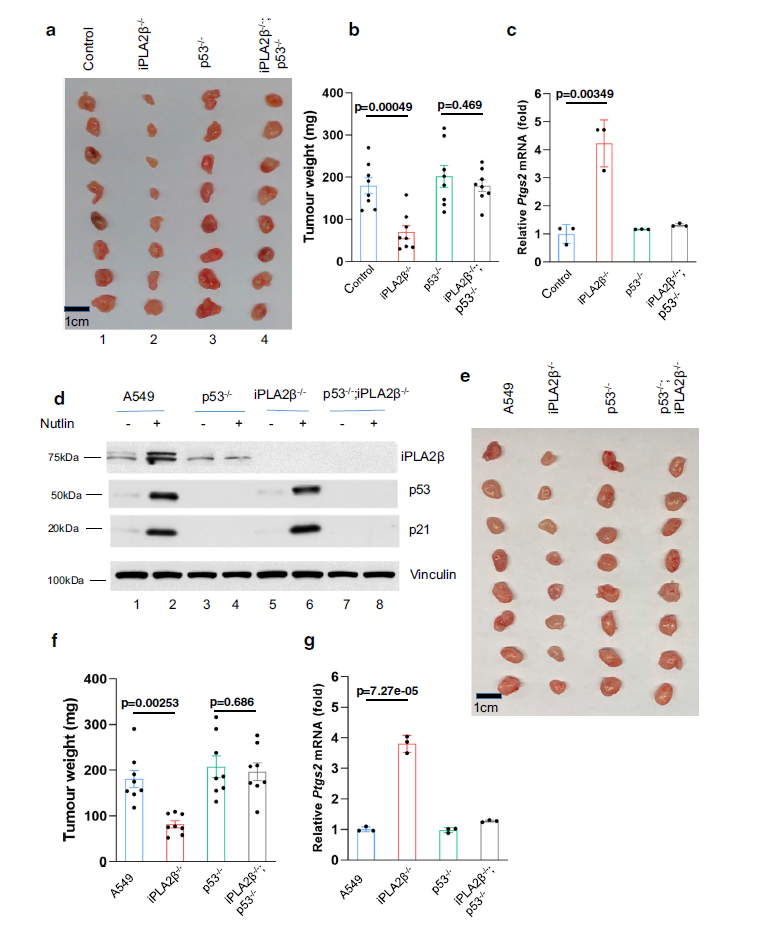
图6 iPLA2β作为p53介导的肿瘤抑制因子的主要抑制因子
4. iPLA2β下调ROS诱导的过氧化物膜脂水平,有效抑制p53诱导的ferroptosis
将ALOX12单独或同时编码ALOX12和iPLA2β的表达载体转染U2OS细胞(图7a),并用C11-BODIPY染色,并通过流式细胞仪检测细胞膜内源性脂质过氧化水平。如图7b、c所示,ALOX12表达显著提高了过氧化物脂质水平;然而,在iPLA2β共表达时,这些升高的水平有效地降低了。由于p53介导的铁下垂独立于GPX4(图1d),作者检测了iPLA2β是否也能在GPX4缺失的细胞中调节p53介导的ferroptosis。为此,我们用编码iPLA2β或iPLA2γ的表达载体转染ACSL4/ GPX4缺失的U2OS细胞(图7d),并用C11-BODIPY染色的流式细胞仪分析脂质过氧化水平。如图7e所示,在ACSL4/ GPX4缺失细胞中,ROS胁迫下p53激活引起的脂质过氧化水平升高(I)通过表达iPLA2β (II)显著降低,但iPLA2γ表达没有降低(III,图7e)。p53-介导GPX4缺失的细胞中,iPLA2β的过表达完全消除了ferroptosis,而iPLA2γ的表达却不能消除ferroptosis(图7f)。因此,iPLA2β通过不依赖GPX4的方式降低脂质过氧化水平,从而抑制p53介导的ferroptosis。
接下来,进行了靶向脂质组学,监测由iPLA2β调节的过氧化物脂质的水平。事实上,水平的氧化磷脂酰乙醇胺(PE)和磷脂酰胆碱(PC) (oxPE (18:0/22:4) sn2或oxPC (18:0/20:4) sn2)显著诱导ALOX12表达式,表明ALOX12在人癌细胞中生成氧化PE和氧化PC中起关键作用, 然而,氧化磷脂酰乙醇胺(PE)和磷脂酰胆碱(PC) (oxPE (18:0/22:4)sn2或oxPC (18:0/20:4)sn2)的水平在iPLA2β表达后显著降低,但不包括酶缺陷突变体iPLA2β (G517C)(图7 g, h)。与这些观察结果一致的是,这种酶缺陷突变体iPLA2β (G517C)也未能抑制p53介导的ferroptosis (图7i)。这些数据表明,ALOX12和iPLA2β在调控氧化磷脂酰乙醇胺(PE)和磷脂酰胆碱(PC)的水平中发挥了关键作用。
在FSP1缺失的细胞中,p53介导的ferroptosis很容易被诱导,更重要的是,在这些细胞中,iPLA2β有效地抑制了细胞死亡(图8a, b)。iPLA2β在正常内稳态条件下的ferroptosis反应中不是必需的,但在应激反应中对控制脂质过氧化水平和ferroptosis至关重要(图8c)。表达较高水平iPLA2β的癌症患者的总生存期明显较短(图8d, e)。综上所述,这些数据表明iPLA2β介导的作用与GPX4或FSP1完全无关。
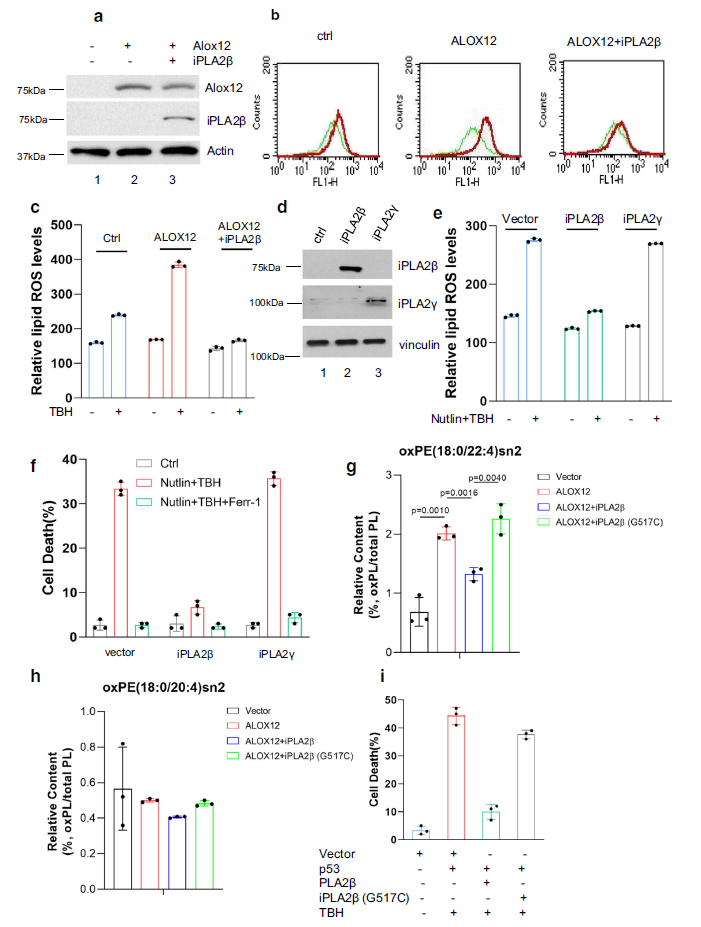
图7 iPLA2β介导抑制p53和ROS应激诱导的脂质过氧化和ferroptosis的机制
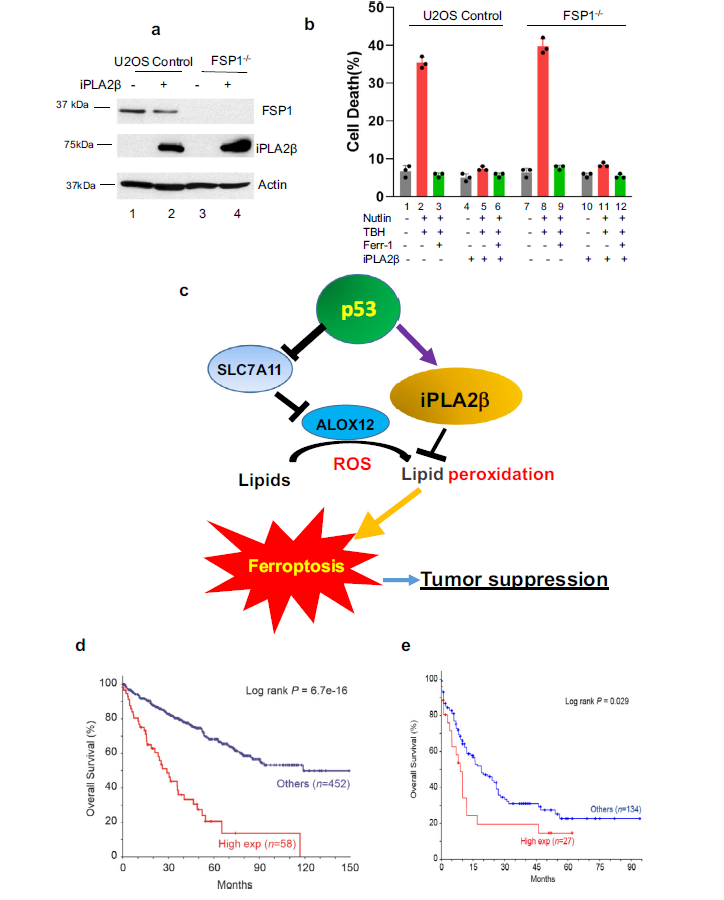
图8 ALOX12和iPLA2β在p53介导的ferroptosis调控中的作用
总结:
研究表明,iPLA2β是激活ferroptosis介导的肿瘤抑制的一个有前途的治疗靶点,且不存在严重的毒性问题。
参考文献:
Chen D, Chu B, Yang X, Liu Z, Jin Y, Kon N, Rabadan R, Jiang X, Stockwell BR, Gu W. iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat Commun. 2021, 12(1):3644. doi: 10.1038/s41467-021-23902-6. PMID: 34131139; PMCID: PMC8206155.