






m6A调控的lncRNA LCAT3在肺癌中发挥致癌作用

lncRNA是重要的表观遗传调控因子,在多种生理和病理过程中发挥着重要作用。然而,lncRNA在肺癌发生中的调控机制尚不清楚。2021年7月发表于“Journal of Hematology & Oncology”(IF=17.388)的文章“LCAT3, a novel m6A‑regulated long non‑coding RNA, plays an oncogenic role in lung cancer via binding with FUBP1 to activate c‑MYC”介绍了一种新的致癌lncRNA-LCAT3在肺癌发生中的调控机制。我们通过分析TCGA中肺癌组织的RNA-seq数据预测并验证了LCAT3。通过甲基化RNA免疫沉淀来评估m6A对LCAT3的修饰。LCAT3-FUBP1-MYC轴通过双荧光素酶报告、RNA免疫沉淀和染色质免疫沉淀检测被评估。利用RNA-seq技术鉴定了LCAT3基因敲低改变的信号通路。此外,通过体内和体外的功能丧失和功能获得试验,研究了LCAT3的作用机制。结果表明,LCAT3在肺腺癌(LUAD)中表达上调,其过表达与LUAD患者的不良预后相关。LCAT3上调可归因于甲基转移酶3 (METTTL3)介导的m6A修饰,导致LCAT3稳定。生物学上,功能缺失分析显示,在体外,LCAT3敲低显著抑制肺癌细胞增殖、迁移和侵袭,在体内,抑制肿瘤生长和转移。LCAT3敲低诱导细胞周期阻滞在G1期。在机制上,LCAT3将FUBP1招募到MYC的FUSE序列上从而激活MYC转录,促进肺癌细胞的增殖、存活、侵袭和转移。
技术路线

结果:
1)LCAT3在肺癌中上调,与预后不良呈正相关
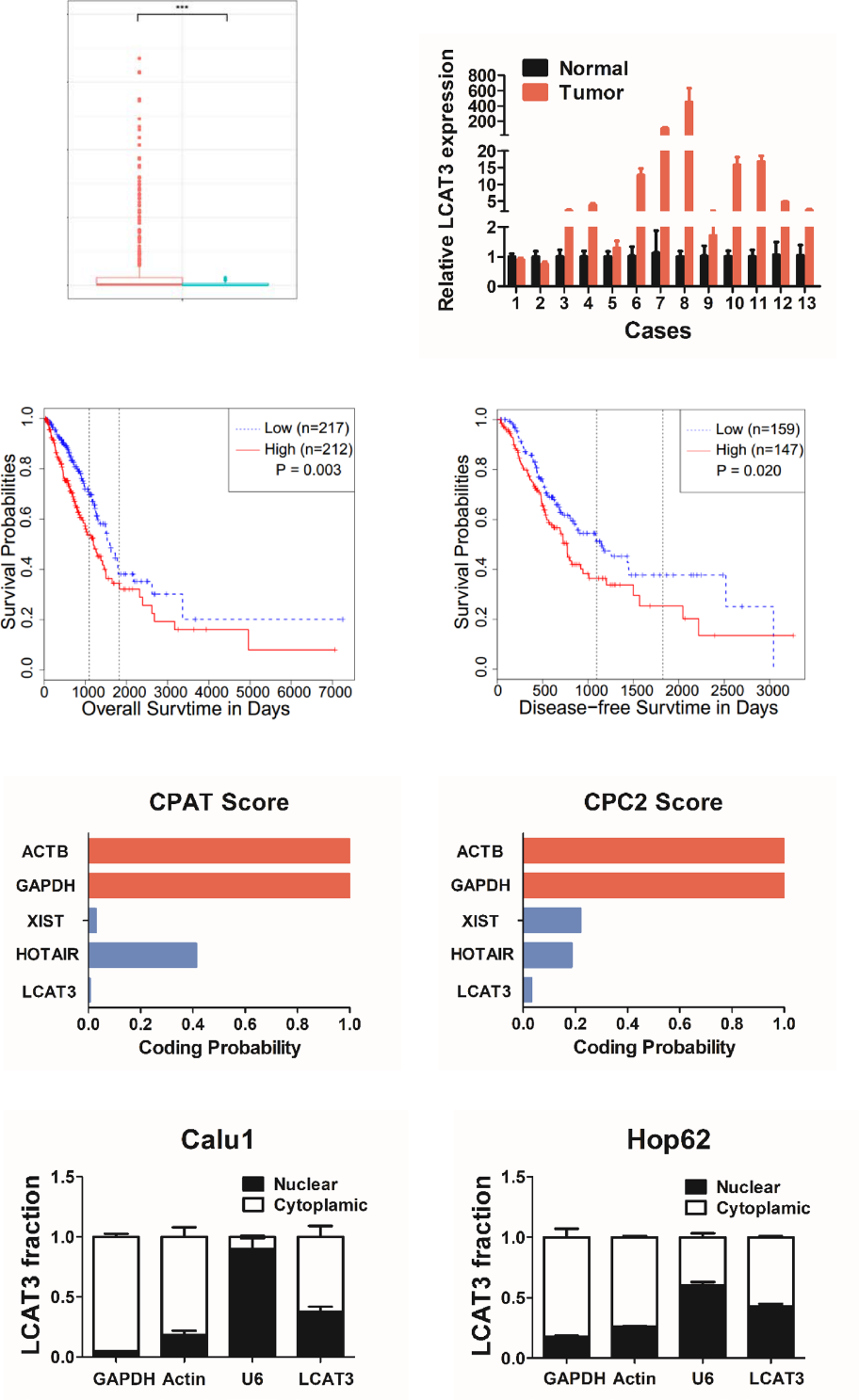
在本研究中,我们研究了一种新的lncRNA,命名为LCAT3。TCGA的RNA-seq数据分析表明,与相邻正常组织相比,LCAT3在LUAD组织中显著上调(图1A)。我们进一步用qRT-PCR验证了LCAT3在13对肺癌样本和相应的非癌肺样本中的表达。与RNA-seq数据一致,qRT-PCR结果证实LCAT3在肺癌组织中上调(图1B)。重要的是,Kaplan-Meier生存分析显示,LCAT3表达水平较高的LUAD患者总生存期和无病生存期均短于LCAT3表达水平较低的LUAD患者(图1C, D)。采用CPAT和CPC2计算LCAT3蛋白编码能力。LCAT3编码蛋白质的潜力低于其他经典的lncRNA,如XIST和HOTAIR(图1E, F),表明LCAT3最有可能是一种lncRNA。亚细胞定位分析显示,LCAT3分布在细胞核和细胞质中(图1G, H)。
2)METTTL3是肺癌中LCAT3上调的原因
m6A修饰是mRNA和lncRNA最普遍的转录后修饰,并调节其翻译、稳定性等。我们的生物信息学分析显示,核心m6A甲基转移酶METTTL3确实在LUAD中上调(图2A)。因此,我们使用CRISPR/Cas9系统敲除肺癌细胞中的METTTL3,western bolt证实了敲除效率(图2B)。我们发现METTTL3的耗竭明显降低LCAT3表达水平(图2C, D)。此外,MeRIP-seq显示在A549细胞中,LCAT3上m6A修饰富集,METTL3敲除后m6A水平降低(图2E)。随后MeRIP-qPCR证实METTL3敲除显著降低了LCAT3 m6A修饰水平(图2F)。最后,我们用放线菌素D处理肺癌细胞以阻断转录,发现METTTL3敲低显著降低LCTA3转录本的半衰期(图2G, H)。总之,METTTL3介导的m6A似乎是LUAD中LCAT3上调的原因,可能是通过稳定其转录本。

3)LCAT3在体内和体外均可促进肺癌细胞增殖
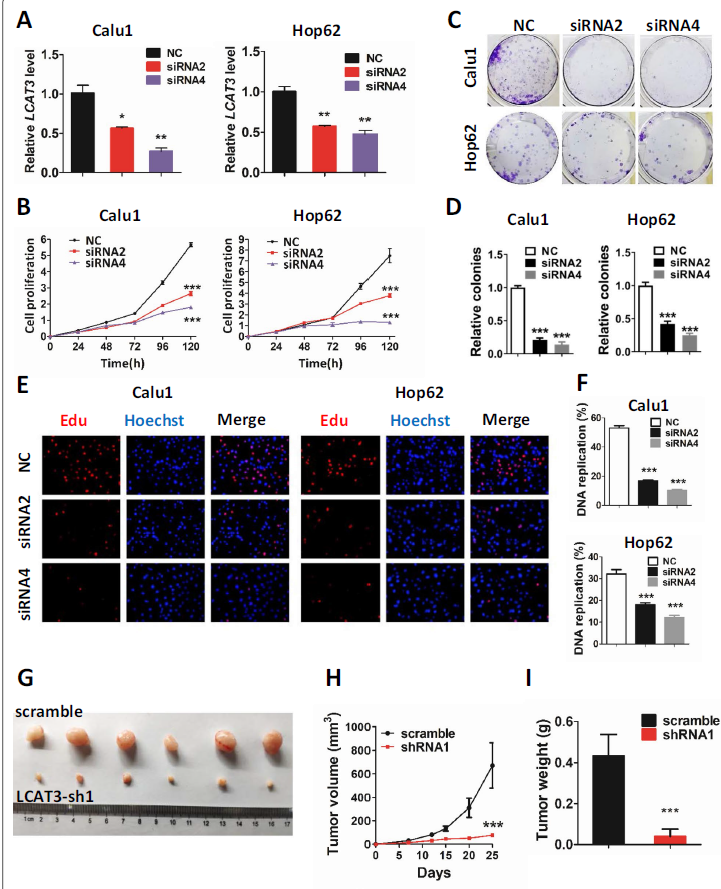
为了研究LCAT3在肺癌细胞中的生物学功能,我们使用siRNA沉默了Calu1和Hop62肺癌细胞中的LCAT3,这充分降低了LCAT3的表达(图3A)。CCK-8检测显示,LCAT3沉默后,细胞增殖明显受到抑制(图3B)。同样,集落形成分析也显示,与siRNA-NC相比,LCAT3敲除可显著减少细胞集落(图3C, D)。此外,EdU分析显示,LCAT3敲除可显著减少肺癌细胞中的DNA复制(图3E, F)。为了进一步评估LCAT3在体内的致癌作用,我们用shRNA稳定敲除Calu1和Hop62细胞中的LCAT3,并通过皮下注射建立移植瘤小鼠模型(图3G)。与shRNA NC组相比,LCAT3敲低组的肿瘤体积和肿瘤重量显著降低(图3H, I)。总体而言,LCAT3对体外和体内肺癌细胞的生长和生存都至关重要。
4)LCAT3对肺癌细胞迁移、侵袭和细胞周期进展的影响
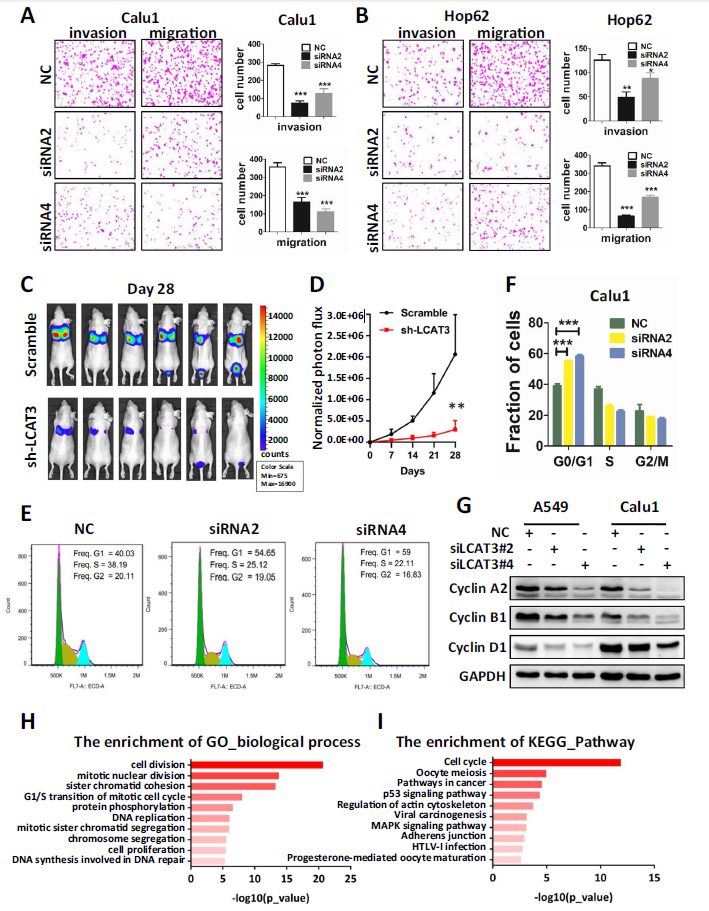
我们探讨了LCAT3对肺癌细胞迁移和侵袭的影响。Transwell分析显示,LCAT3敲低抑制了Calu1和Hop62细胞的迁移和侵袭(图4A, B)。为了探究LCAT3是否也在体内调节肺癌转移,我们将荧光素标记的对照或LCAT3沉默的Calu1细胞静脉注射到裸鼠体内,然后每周对裸鼠进行一次生物发光成像(BLI)。持续的BLI监测显示,4周后注射LCAT3沉默的Calu1细胞的小鼠肺部转移性生长显著下降(图4C, D)。为确定LCAT3基因敲除是否影响肺癌细胞周期,采用流式细胞术检测LCAT3基因敲除和NC肺癌细胞。LCAT3敲低导致G1阻滞(图4E, F)。Western blot检测证实,一些细胞周期相关蛋白,如cyclin A2, cyclin B1和cyclin D1在LCAT3敲低后明显下调(图4G)。为了进一步分析LCAT3对肺癌的影响,我们对LCAT3敲除的肺癌细胞进行了RNA-seq。KEGG通路和GO富集分析显示最显著的KEGG通路和GO项是与细胞周期和细胞分裂相关的,这表明LCAT3沉默导致了细胞周期调控基因的显著改变(图4H, I)。
5)LCAT3与FUBP1发生物理作用,FUBP1在LUAD中也过表达
为了阐明LCAT3在肺癌细胞中作用的潜在分子机制,我们进行了RNA下拉试验来鉴定LCAT3结合蛋白。从凝胶中剪切LCAT3义和反义之间的明显波段用于质谱分析(图5A)。FUBP1因其与LCAT3的特异性结合而被选择进行进一步研究(图5B)。此外,采用抗FUBP1抗体进行RIP检测,进一步证明FUBP1与LCAT3之间的相互作用(图5C)。与FUBP1结合的核心序列为LCAT3的208-342 nt,形成茎环结构。因此,我们使用LCAT3的208-342 nt片段进行RNA拉下分析。结果证实该茎环区(208-342 nt)确实负责LCAT3和FUBP1之间的结合(图5D)。前人研究表明,FUBP1可以分为三个结构域,分别是抑制结构域、DNA和RNA结合结构域和转录结构域。因此,我们构建了flag标记的全长野生型FUBP1突变体的类型和三个缺失域突变体(图5E)。RIP实验显示,LCAT3主要结合于DNA和RNA结合区域(100-447aa)(图5F)。综上所述,这些数据表明LCAT3与FUBP1发生物理作用,LCAT3的茎环区(208-342 nt)和FUBP1的DNA和RNA结合域(100-447aa)是LCAT3与FUBP1结合所必需的。此外,我们发现,FUBP1的mRNA和蛋白水平在LUAD中均显著上调(图5G, H),且在肺癌患者中,FUBP1的高表达与较短的生存时间相关(图5I)。

6)FUBP1沉默可抑制肺癌细胞的增殖和存活
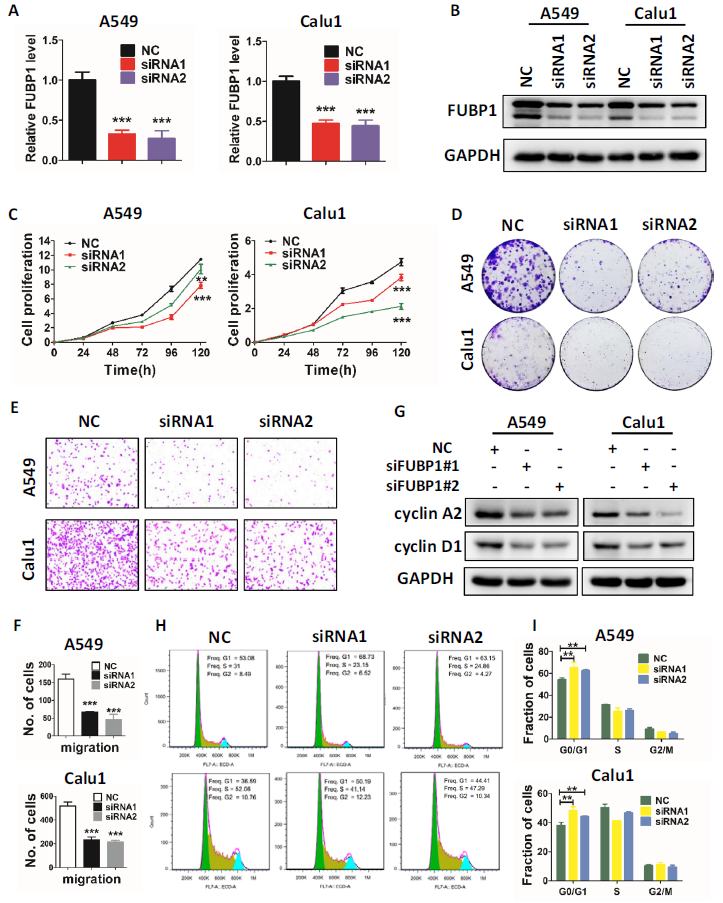
FUBP1在LUAD中的生物学功能尚未被报道,因此,我们通过siRNA敲除A549和Calu1细胞中的FUBP1来评估肿瘤细胞表型的潜在变化。在FUBP1敲低(图6A, B)时,我们观察到肺癌细胞的生长(图6C)和菌落存活(图6D)受到显著抑制。此外,与LCAT3敲低一样,FUBP1敲低也抑制A549和Calu1肺癌细胞的迁移(图6E, F),并通过显著降低cyclin A2和cyclin D1水平触发G1阻滞(图6H, I)(图6G)。总的来说,FUBP1通过增强肺癌细胞的恶性表型发挥致癌作用。
7)LCAT3和FUBP1共同调控c-Myc的表达
我们假设LCAT3将与FUBP1合作调节MYC表达。结果表明,FUBP1的敲除显著抑制了c-MYC mRNA和蛋白质的水平(图7 A, B)。 LCAT3敲除也显著降低c-MYC mRNA和蛋白的水平在A549和Calu1细胞(图7C, D)。因此,与FUBP1一样,LCAT3也是一个c-MYC阳性调节因子。有趣的是,虽然LCAT3或FUBP1的敲低降低了c-MYC的水平,但同时敲低LCAT3和FUBP1并不能进一步降低c-MYC的水平(图7E, F),这表明LCAT3和FUBP1在同一调控点调控c-MYC的表达。
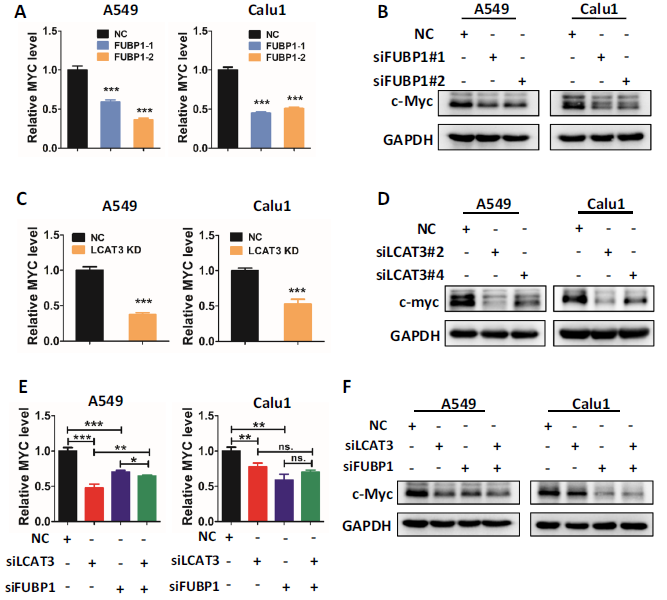
8)LCAT3招募FUBP1激活MYC转录
芯片分析表明,FUBP1在肺癌细胞中与c-MYC启动子结合(图8A)。FUBP1在LCAT3敲低后显著降低(图8 B, C)。因此,LCAT3似乎是FUBP1与c-MYC启动子结合所必需的。然后,然后,我们测试了LCAT3是否通过将FUBP1招募到c-MYC启动子上的FUSE序列中来激活c-MYC表达。使用c-MYC启动子驱动的荧光素酶报告基因,在有或没有FUSE序列的情况下(图8D),我们发现LCAT3或FUBP1基因敲低后,c-MYC启动子活性受到抑制,这种抑制方式依赖于FUSE序列(图8E, F)。因此,LCAT3将FUBP1招募到MYC FUSE序列中来激活其转录。
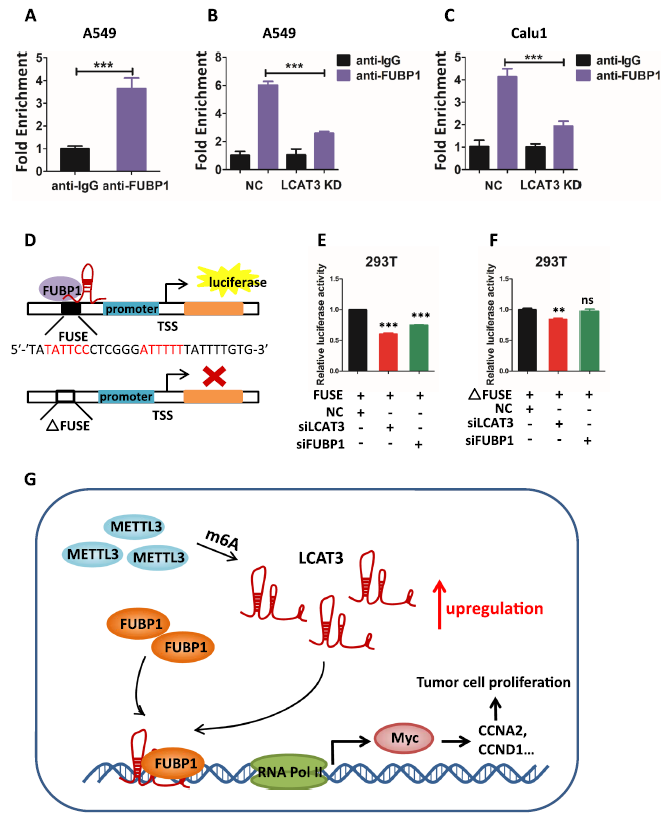
结论:我们鉴定并表征了肺癌中m6A调控的新lncRNA -LCAT3。METTTL3通过m6A修饰稳定LCAT3。过表达的LCAT3将FUBP1招募到c-MYC启动子上,以激活c-MYC表达,导致肺癌细胞增殖、存活、迁移/侵袭和转移增强。我们的研究揭示了LCAT3-FUBP1-cMYC轴的致癌作用,并将其描述为一个有前景的预后生物标志物和潜在的肺癌治疗靶点。