






STK39通过磷酸化增加SNAI1活性促进乳腺癌的侵袭和转移
SNAI1被广泛认为是上皮-间充质转化(EMT)的主要驱动因子,与乳腺癌的进展和转移有关。这种恶性前的作用与翻译后修饰密切相关,特别是磷酸化,它控制其蛋白水平和亚细胞定位。虽然SNAI1稳定性的调控涉及多种激酶,但SNAI1在肿瘤中稳定的确切机制仍有待充分阐明。我们的研究表明STK39是SNAI1稳定性的关键中介,并与转移前细胞过程相关,突出了STK39-SNAI1信号轴作为转移性乳腺癌治疗的有希望的治疗靶点。该文于2021年6月发表于Theranostics(IF= 11.556)杂志上。

技术路线

结果
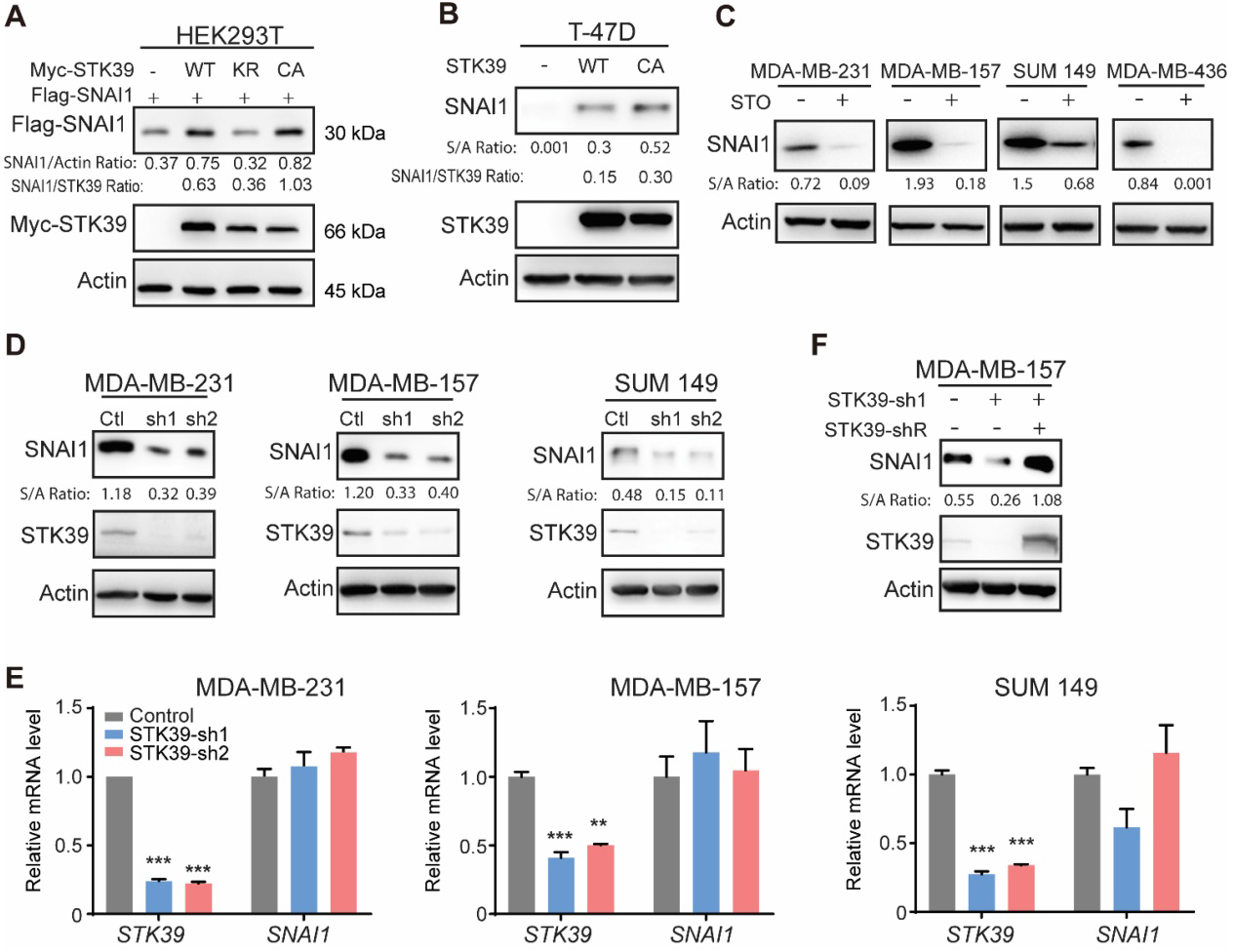
通过质谱分析我们注意到STK39是SNAI1作用的蛋白。为了研究这两个蛋白之间的关系,我们在HEK293T细胞中共同表达了SNAI1和STK39。组成活性突变体STK39激酶(T243E/S383D, CA)显著增加了SNAI1的表达,这表明STK39的酶活性对SNAI1的稳定是必需的(图1A)。STK39 WT和CA也增加了T-47D细胞内源性SNAI1蛋白水平(图1B)。然后我们用STK39抑制剂STOCK2S 26016 (STO)处理几种乳腺癌细胞。STO处理显著降低了SNAI1的表达(图1C)。与此一致的是,在MDA-MB-231、MDA-MB-157和SUM 149细胞中,内源性STK39的下调导致内源性SNAI1蛋白的快速下降,但对mRNA水平没有影响(图1D和1E)。为了排除shRNA的脱靶效应,我们在shRNA介导的MDA-MB-157细胞中用抗shRNA的STK39挽救STK39的表达。如我们所料,STK39的异位表达恢复了SNAI1的表达(图1F)。总之,我们的结果表明STK39稳定SNAI1。
2)STK39通过阻断SNAI1降解来提高SNAI1蛋白的稳定性
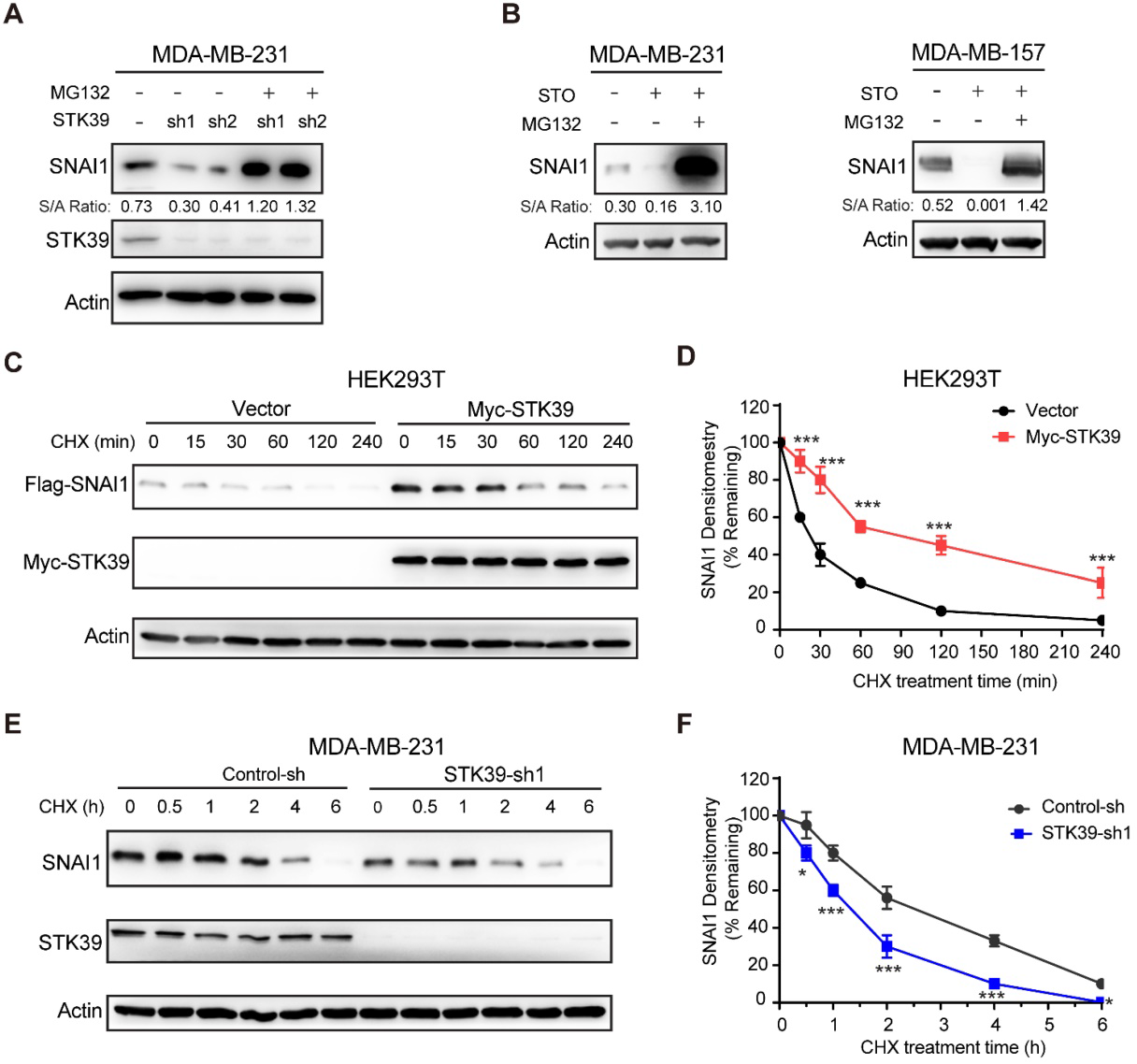
由于SNAI1是一种易降解的蛋白质,容易被蛋白酶体降解,因此我们想知道STK39是否阻断了SNAI1的降解。首先,我们用蛋白酶体抑制剂MG132处理STK39敲低细胞,发现MG132处理后,STK39敲低的MDA-MB-231细胞中SNAI1的下调得以恢复(图2A),说明STK39敲低促进了SNAI1的降解。与此一致的是,MG132处理也恢复了STO处理的MDA-MB-231和MDA-MB-157细胞中SNAI1的表达(图2B)。然后我们检测SNAI1的降解。使用环己亚胺(CHX)阻断新蛋白合成后,SNAI1在转染对照载体的细胞中迅速降解(图2C-2D)。然而,STK39存在时SNAI1水平稳定,而CHX存在时这种效应持续了4 h。为了检测内源性SNAI1是否也受到STK39的类似调控,我们在MDA-MB-231细胞中敲除内源性STK39,发现内源性SNAI1变得不稳定并迅速降解(图2E-2F)。综上所述,这些结果表明STK39通过阻断SNAI1的降解而导致SNAI1的稳定。
3)STK39与SNAI1相互作用,并使T203上的SNAI1磷酸化
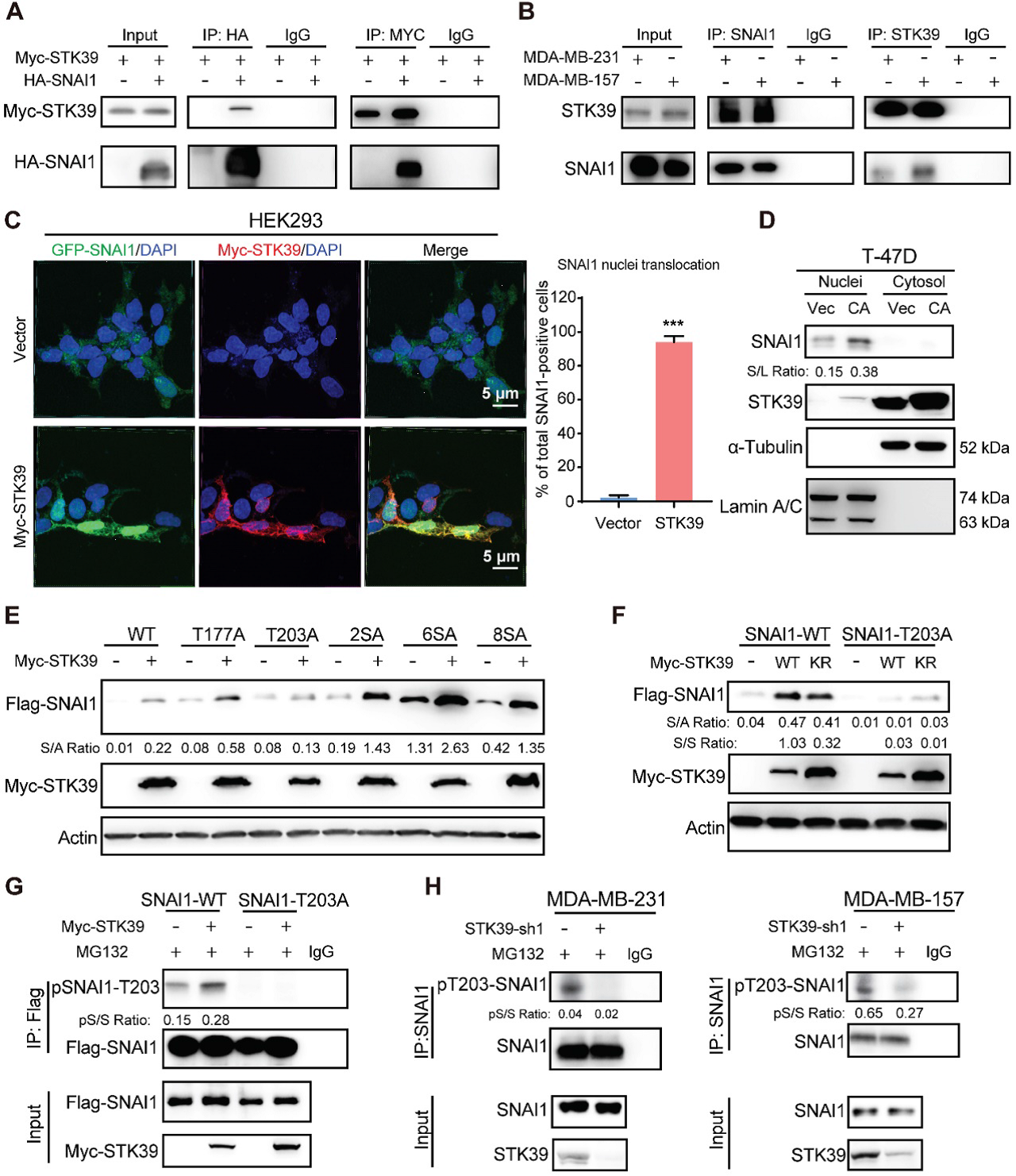
为了描述STK39与SNAI1的相互作用,我们在HEK293T细胞中共表达Myc-STK39和HA-SNAI1,并进行了共同免疫沉淀(IP)实验。在SNAI1的IP之后,我们检测到一个相关的STK39,反之亦然(图3A)。MDA- MB-231和MDA-MB-157细胞中内源性SNAI1和STK39的IP也分别显示内源性STK39和SNAI1的存在(图3B)。然后我们在HEK293细胞中共表达Myc-STK39和GFP-SNAI1。令人惊讶的是,STK39在细胞核中稳定了SNAI1(图3C)。虽然STK39主要定位于胞质,但它包含一个假定的核定位信号,使核转位。由于SNAI1在细胞核内的翻转减少,我们想知道STK39是否会使SNAI1在细胞核内稳定。为了验证这种可能性,我们在T-47D细胞中过表达STK39,并将细胞分成胞质和核部分。SNAI1蛋白仅在细胞核中检测到,在T-47D细胞中SNAI1蛋白的增加主要是在细胞核中(图3D)。然后我们筛选了可能促进SNAI1核保留的丝氨酸/苏氨酸磷酸化位点。结果表明T203是STK39的潜在靶位点(图3E)。与此一致的是,STK39显著提高了WT-SNAI1蛋白水平,但SNAI1-T203A的表达没有显著变化(图3F)。此外,我们在转染野生型SNAI1的HEK293T细胞中检测到SNAI1磷酸化,但未检测到SNAI1 T203A(图3 G)。STK39-WT上调pT203-SNAI1,而STK39-KR不上调pT203-SNAI1。此外,MDA-MB-231和MDA-MB-157细胞中检测到内源性的pT203-SNAI1,而STK39的敲低显著降低内源性的pT203-SNAI1水平(图3H)。这些结果表明STK39介导的T203磷酸化通过细胞核保留导致SNAI1稳定,从而抑制SNAI1降解。
4)STK39以SNAI1依赖的方式增强EMT
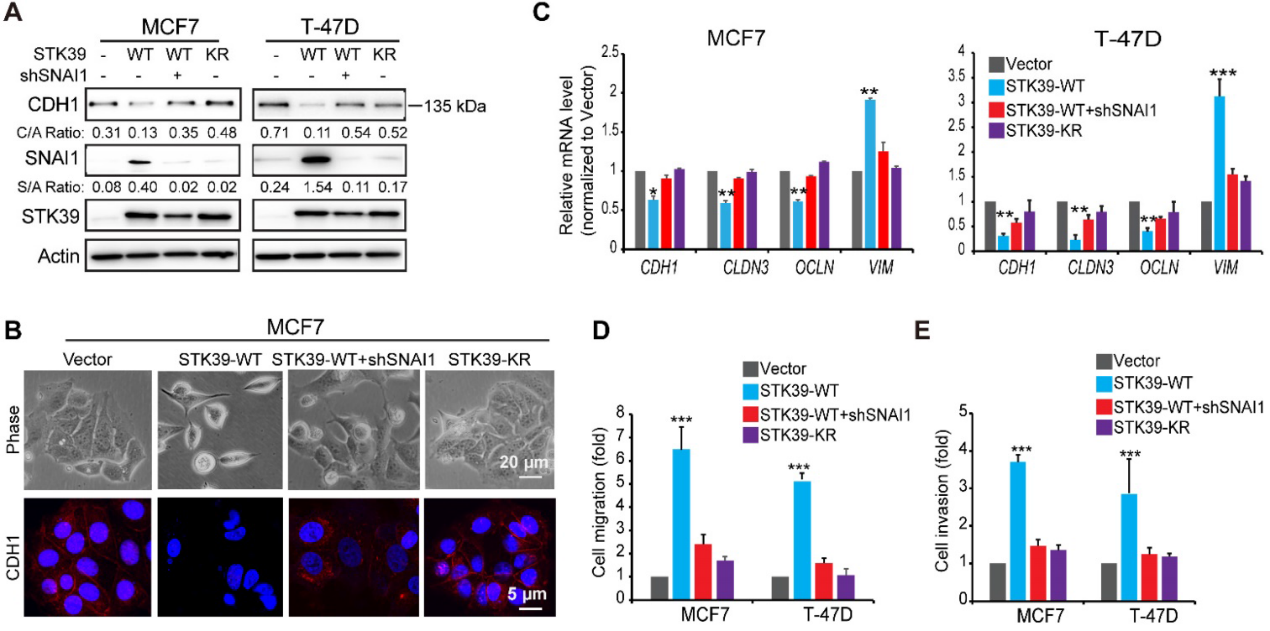
为了探讨STK39的功能作用,我们在两种腔内乳腺癌细胞系MCF7和T-47D中表达了STK39。STK39的表达诱导了这些细胞中SNAI1的稳定和CDH1的下调(图4A)。一致地,STK39的表达诱导了EMT的形态学改变(图4B),并伴有CDH1的下调。此外,RT-PCR显示STK39表达下调了上皮标记物(CDH1, CLDN3和OCLN),上调了间充质分子Vimentin (VIM)的表达(图4C)。在功能上,STK39的表达显著增强了细胞迁移和侵袭能力(图4D-4E)。这些功能需要STK39的催化活性,因为STK39-KR对这些细胞中的SNAI1表达、形态变化、细胞迁移和侵袭均无影响(图4)。重要的是,SNAI1的下调显著减弱了这些变化(图4),表明STK39促进的功能活动需要SNAI1上调。
5)下调STK39可抑制乳腺癌细胞EMT、迁移和侵袭
为了进一步研究STK39在乳腺癌中的作用,我们在MDA-MB-231和MDA-MB-157细胞中建立了STK39敲低的克隆。我们使用两个独立的shRNA实现了内源性STK39 80-90%的敲除效率(图5A)。在这两个克隆中,STK39敲低增加了CDH1的水平,下调了N-cadherin的表达(图5A)。STK39的缺失显著增加了上皮标记物的mRNA表达水平(图5B)。免疫荧光分析也提示CDH1上调,N-cadherin下调(图5C)。STK39基因敲除极大地抑制了这些细胞的迁移和侵袭能力(图5D-5E)。STK39敲除的克隆中SNAI1的表达在很大程度上恢复了STK39消融诱导的效应(图5)。此外,TGF-β1处理诱导MCF10A细胞EMT并激活SNAI1表达(图5F)。STK39缺失显著抑制EMT和SNAI1的表达。综上所述, STK39在很大程度上以SNAI1依赖的方式增强乳腺癌转移。

6)STO表型复制STK39缺陷的影响
STO处理增加了CDH1的表达,下调了SNAI1的表达(图6A),且呈剂量依赖性(图6B)。STO处理还上调了CDH1的水平,下调了N-cadherin的表达(图6C)。免疫荧光分析进一步显示CDH1升高,N-cadherin降低(图6D)。与STK39缺陷相一致,STO处理可上调上皮标记物(图6E),并极大地抑制这些细胞的迁移和侵袭(图6F-6G)。总之,用STK39抑制剂处理后,观察到STK39表达缺失的效应;具体来说,包括迁移受损、SNAI1下调和CDH1表达增加,因此支持STK39激酶活性在EMT中发挥关键作用。

7)在体内抑制STK39可使化疗增敏并阻断转移
SNAI1与获得耐药相关。紫杉醇(PTX)是经典的紫杉烷类药物,用于乳腺癌治疗。不幸的是,对患者有效和成功的治疗通常受到获得性耐药性的限制。为了检测STK39的下调是否会提高PTX的敏感性,我们测定了在STK39缺失或不缺失的情况下,PTX对MDA-MB-231和MDA-MB-157细胞株的IC50。STK39的缺失降低了PTX的IC50(图7A)。为了确定STO是否与PTX有协同作用,我们用PTX处理MDA-MB-231和MDA-MB-157细胞。STO与PTX协同抑制细胞增殖(图7B)。软琼脂菌落形成分析显示,与PTX单独处理相比,STO和PTX联合处理在菌落形成和菌落大小上都有更大的降低(图7C)。我们的数据表明,STK39抑制使乳腺癌细胞对PTX敏感。接着,为了直接评估STK39在体内是否对细胞转移至关重要,我们将STK39基因敲低的MDA-MB-231-荧光素酶细胞静脉注射到雌性SCID小鼠中,并对这些小鼠进行生物发光成像(BLI)。所有的对照组小鼠由于大量肺转移而死亡(图7D)。相比之下,注射了STK39敲除细胞的小鼠是活的,没有检测到转移。组织学分析显示,对照细胞有大量转移灶,而STK39敲除细胞缺乏转移灶(图7E-7F)。与体外SNAI1的功能一致,外源性SNAI1在STK39敲除细胞中的表达在很大程度上挽救了肺转移的形成(图7D-7F)。在乳腺癌中,SNAI1表达与无瘤生存率呈负相关。为了验证表达高水平STK39的原发性乳腺癌患者是否比表达低水平STK39的乳腺癌患者复发更快,我们分析了两个来自原发人乳腺癌的微阵列表达数据集。结果发现STK39高表达的个体的总生存率或无病生存间隔降低(图7G)。这些结果表明STK39的表达可能是临床环境中乳腺癌的重要预后指标。

结论:我们的研究揭示了STK39通过增强SNAI1的稳定性促进肿瘤细胞EMT和转移的机制。我们的研究也对STK39靶向治疗转移性癌症具有重要意义。