






维生素D-维生素D受体调控糖尿病小鼠肾小管上皮细胞的缺陷自噬
糖尿病肾病(DN)已成为终末期肾病的主要病因之一,自噬障碍与DN的发病机制有关。我们前期研究发现维生素D (VD)和VDR(维生素D受体)通过抑制炎症和纤维化发挥肾保护作用。然而,VD-VDR是否调节DN患者的自噬障碍尚不清楚。在本研究中,我们建立了链脲佐菌素(STZ)诱导的VDR敲除(VDR-KO)小鼠和VDR特异性过表达(VDR-OE)小鼠的糖尿病模型。结果表明,paricalcitol(一种激活的维生素D类似物)或VDR-OE均能减轻STZ诱导的白蛋白排泄、肾小管损伤和炎症,而VDR-KO小鼠的这些症状均加重。STZ小鼠肾脏存在自噬缺陷,VDR-KO小鼠的自噬缺陷更明显,paricalcitol或VDR-OE可部分修复。在高糖诱导的HK-2细胞中,观察到自噬缺陷和PRKAA1/AMPK磷酸化降低,paricalcitol可通过VDR依赖的方式部分恢复PRKAA1/AMPK磷酸化。AMPK抑制剂可消除paricalcitol诱导的自噬激活,AMPK激活剂可修复高糖诱导的HK-2细胞的缺陷自噬。此外,paricalcitol介导的AMPK激活可被CAMKK2/CaMKKβ抑制消除,而STK11/LKB1敲除不能。同时,paricalcitol可以挽救高糖诱导的Ca2+浓度下降。该文于2021年8月发表于Autophagy(IF= 16.016)杂志上。

技术路线
结果
1)VD-VDR延迟DN进展
诱导12周后,STZ诱导的糖尿病WT小鼠出现蛋白尿,而pari治疗阻断了STZ诱导小鼠的蛋白尿,而不改变血糖水平或体重。同时,在STZ诱导下,VDR-KO小鼠比WT小鼠出现更严重的蛋白尿(图1A)。PAS染色显示STZ治疗后肾小管不同程度的部分扩张,近端肾小管上皮细胞(PTECs)扁平,刷状边界减少,VDR-KO进一步恶化,pari治疗部分阻止(图1B)。另一方面,VDR-OE减少了STZ诱导的蛋白尿,而不影响体重和血糖水平(图1C)。PAS染色显示WT+STZ和OE+STZ小鼠近端小管上皮细胞均不同程度扁平,OE+STZ小鼠表现出部分减弱的PAS阳性染色和管状损伤(图1D)。

2)VD-VDR可减轻STZ诱导的糖尿病小鼠肾脏炎症
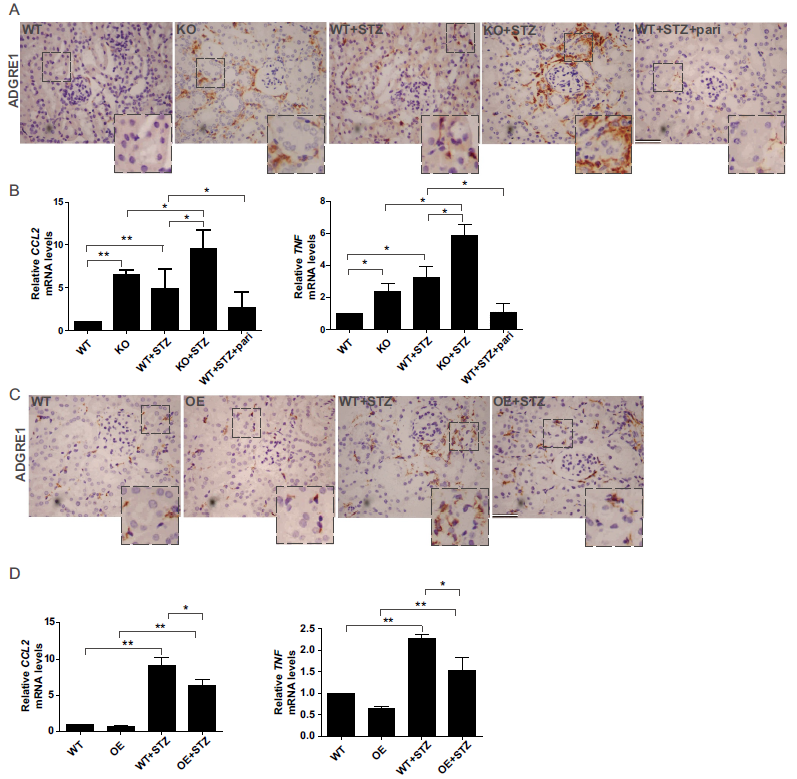
由于炎症在DN的发展中起重要作用,我们评估了VD-VDR对STZ诱导的糖尿病小鼠炎症的影响。巨噬细胞浸润标志物ADGRE1/F4/80在VDR-KO小鼠和STZ诱导的糖尿病WT小鼠中升高。ADGRE1在KO+STZ小鼠中增长最为显著,在WT+STZ+pari组中明显受到抑制(图2A)。RT-PCR显示,VDR-KO小鼠和STZ诱导的糖尿病小鼠中促炎细胞因子(CCL2/MCP-1和TNF/TNF- α)的表达明显高于WT小鼠。这种增长在KO+STZ小鼠中最为强劲。此外,pari部分恢复了促炎细胞因子的增加(图2B)。与WT和VDR-OE小鼠相比,STZ诱导的糖尿病WT小鼠ADGRE1、CCL2和TNF的表达均增加。VDR过表达明显降低了炎症浸润,OE+STZ小鼠的炎症因子表达低于WT+STZ小鼠(图2C,D)。
3)VD-VDR可缓解糖尿病小鼠肾脏异常自噬体积聚
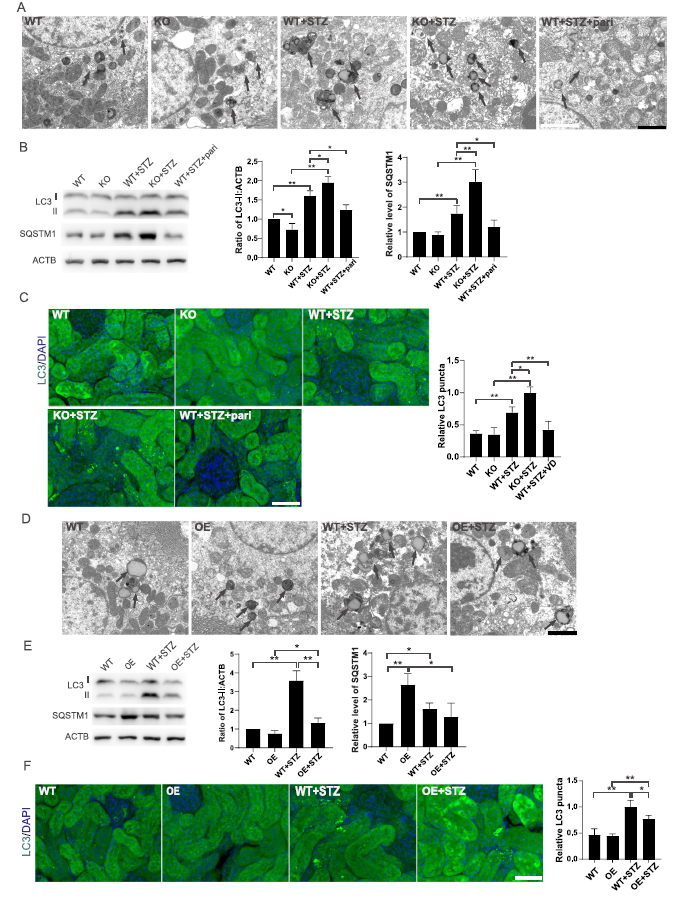
我们利用透射电子显微镜(TEM)检查糖尿病小鼠肾脏的变化。与WT小鼠相比,WT+STZ小鼠肾小管上皮细胞中发现更多的自噬空泡,糖尿病VDR-KO小鼠中发现最多的自噬空泡(图3A,D)。为了进一步证实这些发现,我们检测了自噬的关键标记LC3的表达。如图3B和3E所示,STZ处理后小鼠LC3- II增加,KO+STZ组这种作用更明显,pari处理恢复了STZ诱导的LC3变化。为了进一步验证,我们对自噬体进行了免疫荧光染色。STZ小鼠LC3斑点增多,KO+STZ小鼠LC3斑点增多更为明显,pari处理小鼠LC3斑点减少(图3C,F)。为了探究自噬空泡和LC3数量的增加是否表明自噬激活或自噬降解受损,我们检测了自噬底物SQSTM1。如图3B和3E所示,STZ诱导的糖尿病小鼠的SQSTM1表达高于WT小鼠,表明STZ诱导的小鼠存在自噬缺陷。Pari部分恢复了缺陷自噬,因为它降低了STZ诱导的糖尿病小鼠中SQSTM1的表达。此外,STZ处理后VDR-KO小鼠的自噬缺陷水平高于对照小鼠,而OE+STZ小鼠未见SQSTM1聚集。我们的数据表明,STZ诱导的糖尿病肾脏的自噬是有缺陷的,可以通过VD-VDR部分恢复。
4)VD可修复高糖诱导的HK-2细胞自噬缺陷
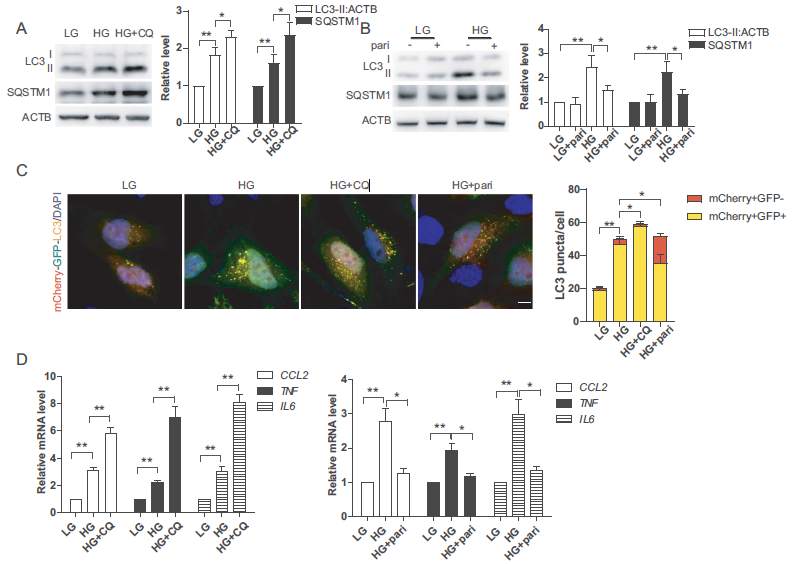
我们探讨了pari对体外DN细胞自噬和炎症的影响。我们用高水平葡萄糖(HG)处理HK-2细胞。如图4A所示,在高糖条件下,LC3-II和SQSTM1的表达增加,在自噬抑制剂氯喹(CQ)作用下,LC3-II和SQSTM1的表达进一步增加。此外,我们使用tf-LC3研究自溶酶体的成熟过程。在酸性溶酶体环境中,mCherry比GFP更稳定,自溶酶体的正常成熟以增加仅红点为特征。与此相反,GFP和mCherry斑点共定位表明自噬通量中断,呈现黄色斑点。高糖诱导mCherry和GFP共定位,CQ处理更明显(图4C)。这些结果进一步表明,高糖可损害自噬通量。另一方面,与HG组相比,paricalcitol降低了LC3-II和SQSTM1的表达,增加了仅红色的斑点(图4B,C)。为了研究缺陷性自噬是否有助于高糖诱导的炎症反应,采用实RT-qPCR检测促炎因子(CCL2, TNF, IL6)的mRNA水平。结果显示CQ加重了高糖诱导的炎症反应,而paricalcitol降低了促炎因子的表达(图4D),说明HK-2细胞高糖诱导的炎症反应部分是由于自噬缺陷引起的,paricalcitol可以恢复自噬通量,减少炎症反应。
5)VD通过激活AMPK通路恢复了缺陷的自噬通量
有报道称,高糖诱导的自噬变化与AMPK密切相关。为了探讨paricalcitol诱导HK-2细胞自噬的分子机制,我们研究了AMPK-ULK1激酶网络的状态。如图5A所示,在高糖条件下,PRKAA1/AMPKα1的磷酸化水平降低,而paricalcitol处理则逆转它的表达。在PRKAA1抑制剂复合物C的存在下,paricalcitol诱导的自噬激活被抑制(图5A)。相反,二甲双胍,PRKAA1激活剂,可降低高糖诱导的LC3-II、SQSTM1和炎症水平(图5B,C)。这些结果表明,AMPK激活是paricalcitol诱导的高糖条件下自噬激活和炎症抑制的关键机制。
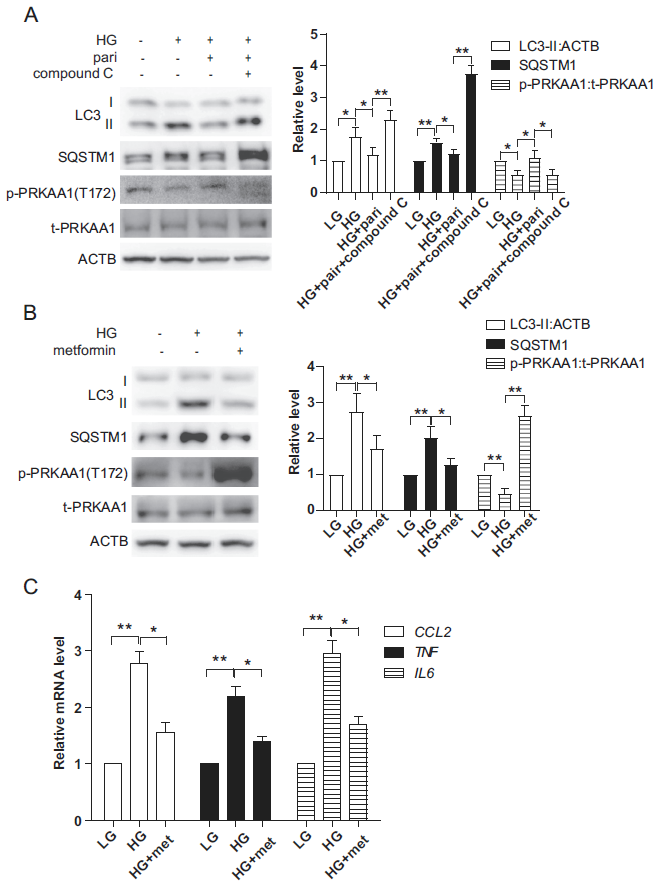
6)VD以VDR依赖的方式调控AMPK
由于维生素D通过VDR发挥其最大的生物学作用,我们使用HK-2 VDR KO细胞研究VD是否通过VDR激活AMPK。结果表明,paricalcitol增加了PRKAA1和ULK1的磷酸化,而在高糖条件下则降低了PRKAA1和ULK1的磷酸化。而在HK-2 VDR KO细胞中,这种作用完全消失(图6A)。此外,二甲双胍在高糖条件下激活了HK-2 WT细胞中的PRKAA1和ULK1,但二甲双胍的这种作用在HK-2 VDR KO细胞中仍然可以观察到(图6B)。这些结果表明VD以VDR依赖的方式激活AMPK。
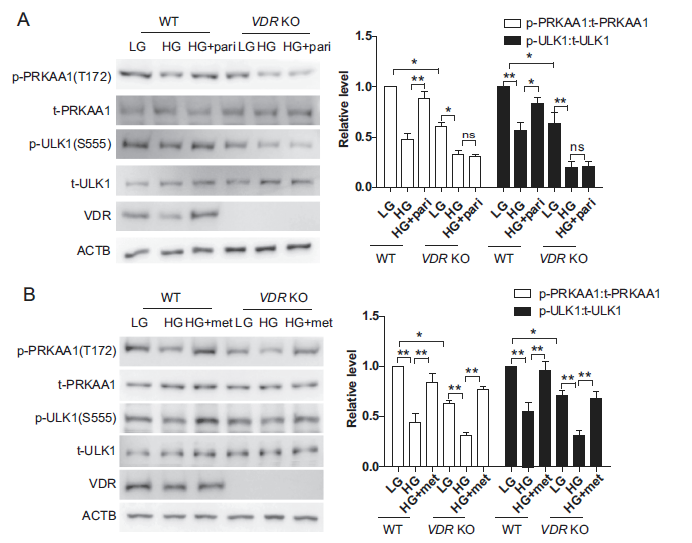
7)VD通过Ca2+-CAMKK2途径调控AMPK
AMPK通过上游激酶磷酸化激活,主要包括STK11/LKB1和CAMKK2/CaMKKβ。为了确定AMPK激酶是否参与paricalcitol介导的AMPK激活,我们使用STO- 609(选择性CAMKK抑制剂)、CAMKK2 siRNA和HK-2 STK11 KO细胞。如图7A和7B所示,与STO-609或CAMKK2 siRNA共同处理完全消除了paricalcitol刺激的PRKAA1和ULK1磷酸化。然而,paricalcitol能够激活HK-2 STK11 KO细胞中的PRKAA1和ULK1(图7C)。这些结果表明paricalcitol通过CAMKK2激活AMPK。由于CAMKK2主要受细胞内Ca2+浓度的调节。为了阐明Ca2+信号在paricalcitol介导作用中的参与,我们使用荧光Ca2+探针Fluo 4AM评估了细胞内Ca2+浓度的变化对paricalcitol处理的响应。如图7D所示,当HK2细胞暴露于高糖环境时,Ca2+浓度下降,paricalcitol可以缓解这种下降。
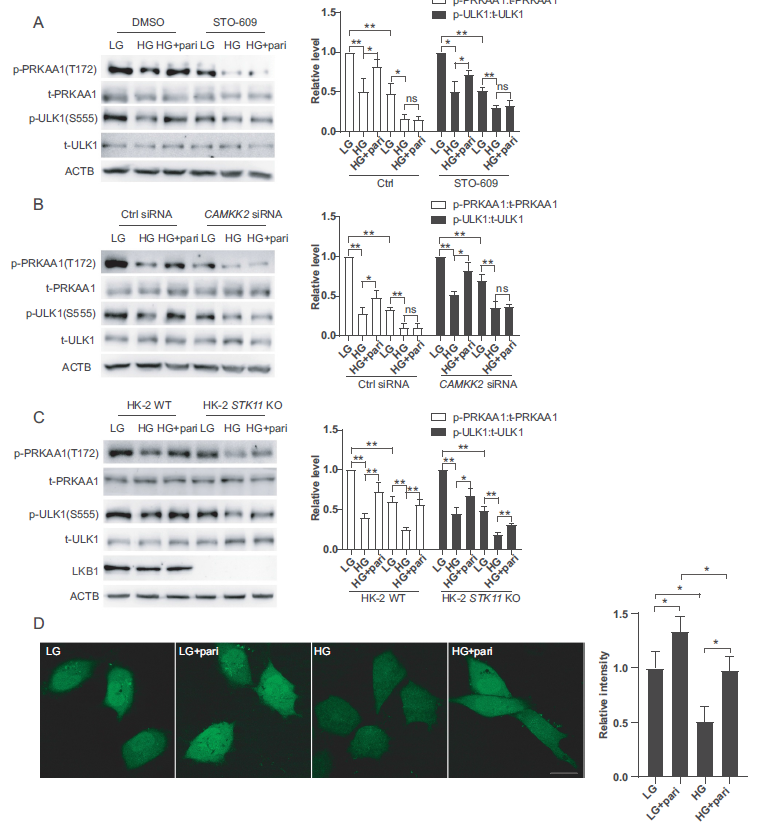
8)VDR激活STZ诱导的糖尿病小鼠AMPK
为了进一步证实VD-VDR对高糖环境下AMPK激活的影响,我们检测了STZ诱导的糖尿病小鼠AMPK- ULK1的磷酸化水平。如图8所示,STZ诱导的糖尿病小鼠PRKAA1和ULK1磷酸化水平降低,KO+STZ小鼠的这种变化更明显。此外,paricalcitol或VDR过表达促进PRKAA1和ULK1磷酸化,尽管WT+STZ组和OE+STZ组之间没有统计学差异(图8A,C)。我们还通过免疫荧光染色检测了p-PRKAA1的水平,其变化趋势与蛋白印迹相似(图8B,D)。结合图3E的结果,我们推测在STZ诱导的糖尿病肾脏中,VDR需要VD激活才能有效磷酸化PRKAA1,然后调节自噬(图3E和8A,B)。综上所述,VD-VDR通过激活AMPK-ULK1通路恢复了stz诱导的糖尿病小鼠肾脏缺陷的自噬。
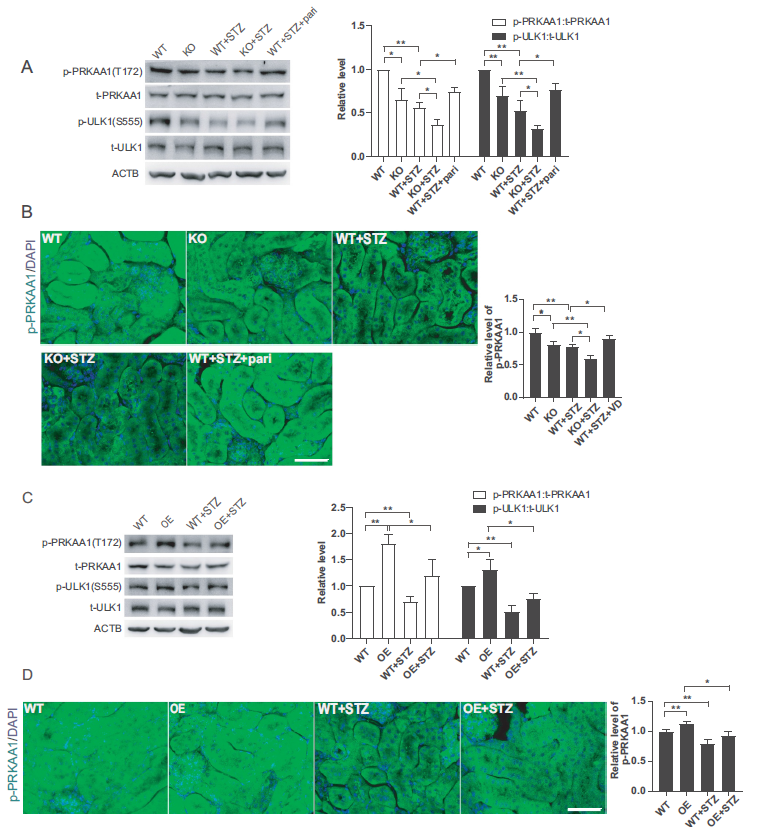
结论:我们的研究结果表明STZ诱导的糖尿病小鼠肾脏中存在缺陷自噬,VD-VDR部分恢复了缺陷自噬并减少了炎症。机制研究表明,AMPK参与了高糖诱导的缺陷自噬和炎症,VD刺激的AMPK磷酸化是由于CAMKK2激活响应细胞内Ca2+浓度增加。我们的结果阐明了VD-VDR在DN中肾保护作用的新机制。