






二甲双胍通过抑制乳腺癌中 SLC7A11 的 UFMylation 诱导铁死亡
铁死亡是一种新定义的受调节细胞死亡形式。靶向铁死亡被认为是一种新的抗癌策略。尽管越来越多的证据表明抗糖尿病二甲双胍作为抗癌剂的潜在功效,但尚未完全阐明这种功效背后的确切机制。我们研究发现二甲双胍以不依赖 AMPK 的方式诱导铁死亡以抑制肿瘤生长。从机制上讲,我们证明二甲双胍增加细胞内 Fe2+ 和脂质 ROS 水平。具体而言,二甲双胍通过抑制其 UFMylation过程来降低 SLC7A11 的蛋白质稳定性。此外,二甲双胍与柳氮磺吡啶系统xc-抑制剂联合使用可以协同作用诱导铁死亡并抑制乳腺癌细胞的增殖。该研究首次证明二甲双胍诱导铁死亡的能力可能是其抗癌作用的一种新机制。此外,我们发现SLC7A11是一种新的UFMylation底物,并发现针对UFM1/SLC7A11通路可能是一种有前景的癌症治疗策略。本文于2021年6月发表在Journal of Experimental & Clinical Cancer Research(IF=11.161)期刊上。

技术路线

结果:
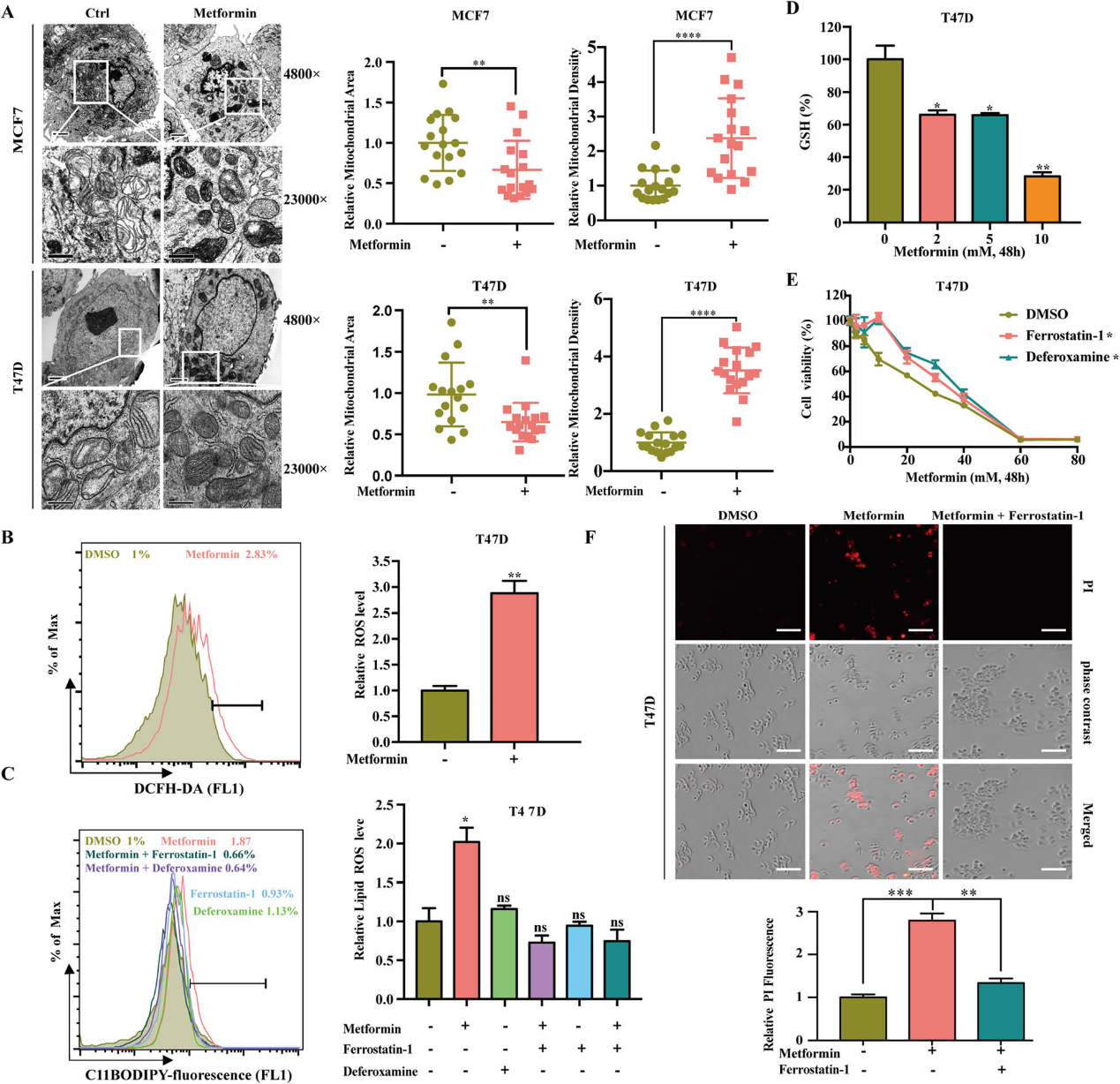
1)二甲双胍触发铁依赖性细胞死亡
越来越多的证据表明二甲双胍在临床预防和治疗乳腺癌方面具有巨大的潜力。我们的研究表明二甲双胍能有效地抑制乳腺癌细胞的增殖,且呈剂量和时间依赖性(图1A)。接下来,我们探讨哪些细胞死亡形式参与二甲双胍在乳腺癌细胞的抗癌活性。我们使用了多种细胞死亡抑制剂,包括Z-VAD-FMK(一种凋亡抑制剂)、necrosulfonamide (一种坏死抑制剂)和3-Methyladenine (3-MA,一种自噬抑制剂)。由于二甲双胍已被证明可以调节细胞中的铁稳态,我们还引入了一种铁清道夫,脱铁胺(DFO)。结果显示DFO恢复了二甲双胍培养的T47D和MCF7细胞的活力(图1B)。Z-VAD-FMK、necrosulfonamide和3-MA也较DMSO组略微提高了细胞存活率(图1 B)。虽然二甲双胍可以诱导细胞凋亡并抑制自噬,但DFO的作用远比凋亡和自噬抑制剂显著(图1B)。此外,二甲双胍促进了铁离子的积累,而DFO抑制了铁离子水平的升高(图1C和D)。碘化丙啶染色证实,DFO抑制了二甲双胍诱导的T47D细胞死亡(图1E)。综上所述,二甲双胍的抗癌作用需要铁离子,而二甲双胍诱导的铁离子增加可能是二甲双胍发挥抗癌作用的机制之一。

2)二甲双胍可引起铁死亡
为了阐明二甲双胍和铁死亡之间的关系,我们首先研究了二甲双胍对铁死亡形态学特征的影响。铁透射电镜显示,二甲双胍诱导MCF7和T47D细胞线粒体体积减小,膜密度增加(图2A)。接下来,我们重点研究了二甲双胍对铁死亡生化过程的影响。我们用不同浓度的二甲双胍处理乳腺癌细胞。研究结果显示,二甲双胍提高了细胞内总ROS和脂质ROS的水平,脂质ROS的增加被ferroptosis抑制剂(DFO, Fer-1)抑制,而DFO和Fer-1本身对其影响不大(图2B和C)。此外,二甲双胍还能降低细胞内谷胱甘肽水平(图2D)。我们还检测了铁死亡抑制剂(DFO, Fer-1)是否可以逆转二甲双胍的抗癌作用。我们的结果表明,二甲双胍处理T47D细胞时,DFO和Fer-1都能显著挽救细胞活力(图2E)。此外,碘化丙啶染色证实,fer1可抑制二甲双胍诱导的T47D细胞死亡(图2f)。综上所述,二甲双胍可引起铁死亡。
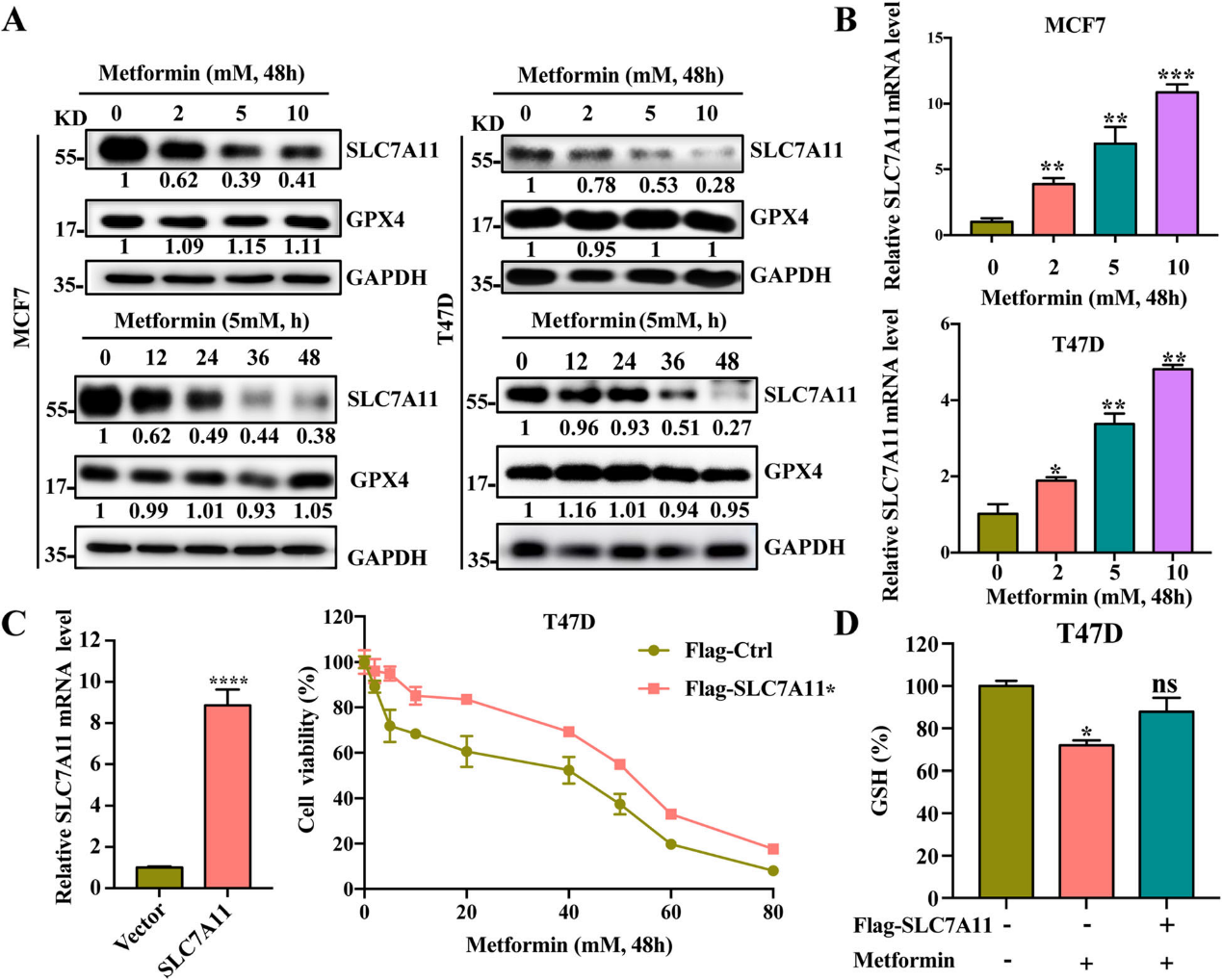
3)二甲双胍抑制SLC7A11的表达
为了明确二甲双胍调控铁死亡的具体分子机制,我们检测了不同二甲双胍浓度和处理时间对SLC7A11和GPX4表达的影响。结果显示,二甲双胍能有效抑制T47D和MCF7细胞中SLC7A11的表达,且呈剂量和时间依赖性,而转录水平显著升高(图3A和B)。但二甲双胍对GPX4的表达无显著影响(图3A)。为了进一步阐明SLC7A11在二甲双胍抗癌作用中的作用,我们在T47D细胞中敲除或过表达SLC7A11。结果表明,SLC7A11过表达显著逆转二甲双胍的抗癌作用和GSH的上调(图3C和D)。这些结果提示二甲双胍可以通过下调SLC7A11蛋白水平而诱导铁死亡抑制癌症。
4)二甲双胍可调节UFM1的表达
UFMylation是一种泛素样修饰,在乳腺癌的发生和发展中起重要作用。通过对DRUGSURV数据库的分析,我们发现二甲双胍可以间接靶向UFM1(图4A)。我们推测UFM1可能在二甲双胍的抗癌作用中发挥一定的作用。结果显示,二甲双胍在一定程度上抑制了UFM1蛋白水平和UFMylation修饰水平(图4B),但不影响UFM1转录水平(图4C)。为进一步阐明UFM1对二甲双胍抑瘤活性的影响,在T47D细胞中过表达UFM1。结果表明,过表达UFM1可有效阻断二甲双胍的抗癌作用,恢复二甲双胍介导的GSH抑制水平(图4 D和E)。综上所述,二甲双胍可能通过UFM1发挥抗癌作用。

5)二甲双胍通过抑制SLC7A11的UFMylation而下调SLC7A11的表达
铁死亡受到SLC7A11的密切调控,因此,探索其调控机制有助于进一步找到诱导铁死亡在癌症治疗中的新的治疗靶点。我们首先检测了SLC7A11和UFM1在不同乳腺癌细胞株中的表达。结果显示,SLC7A11与UFM1蛋白水平之间存在明显的正相关(图5A)。其次,生物信息学分析表明SLC7A11的表达与乳腺癌患者的预后存在显著的负相关,而UFM1的表达与乳腺癌患者的预后没有显著的负相关(图5 B)。为了确定SLC7A11和UFM1是否具有调控作用,我们在敲低UFM1后检测SLC7A11表达的影响。结果表明,抑制UFM1可下调SLC7A11的表达,但不影响其转录水平(图5C)。此外,抑制UFM1抑制了SLC7A11的蛋白稳定性(图5D)。为了确定SLC7A11是否可以被UFM1修饰,我们检测了在UFM1敲低或不敲低情况下SLC7A11的UFMylation。我们发现SLC7A11是UFMylation的底物,其修饰的特异性通过敲除UFM1来确定(图5E)。进一步的UFMylation修饰试验表明,SLC7A11 UFMylation可以被UfSP2抑制(图5F)。这些发现将SLC7A11定义为UFM1的一种新的修饰底物。此外,二甲双胍能有效抑制SLC7A11 UFMylation水平(图5G)。综上所述,二甲双胍通过抑制SLC7A11的UFMylation来下调SLC7A11的蛋白稳定性,从而诱导铁死亡。

6)二甲双胍可诱导不依赖于AMPK途径的铁死亡
目前,许多研究表明二甲双胍可以直接作用于肿瘤细胞,通过AMPK依赖或AMPK不依赖的信号通路发挥其抗肿瘤生物学作用。我们的结果也表明二甲双胍可以激活AMPK磷酸化(图6A)。因此,为了确定AMPK通路是否参与二甲双胍对铁死亡的调控,我们首先明确AMPK的激活是否能诱导铁死亡。结果显示,AICAR,一种AMPK活化剂,增加脂质ROS生产和抑制SLC7A11表达,表明AMPK活化可以诱导铁死亡(图6 E和F)。此外,二甲双胍与化合物C联合作用可协同提高脂质ROS水平,抑制GSH水平和SLC7A11的表达(图6 A, B, C)。化合物C本身可以增加脂质ROS和Fe2+的产生(图6 C和D)。总之,这些发现表明二甲双胍或AMPK失衡均可诱导铁死亡,而二甲双胍可诱导铁死亡而不涉及AMPK。

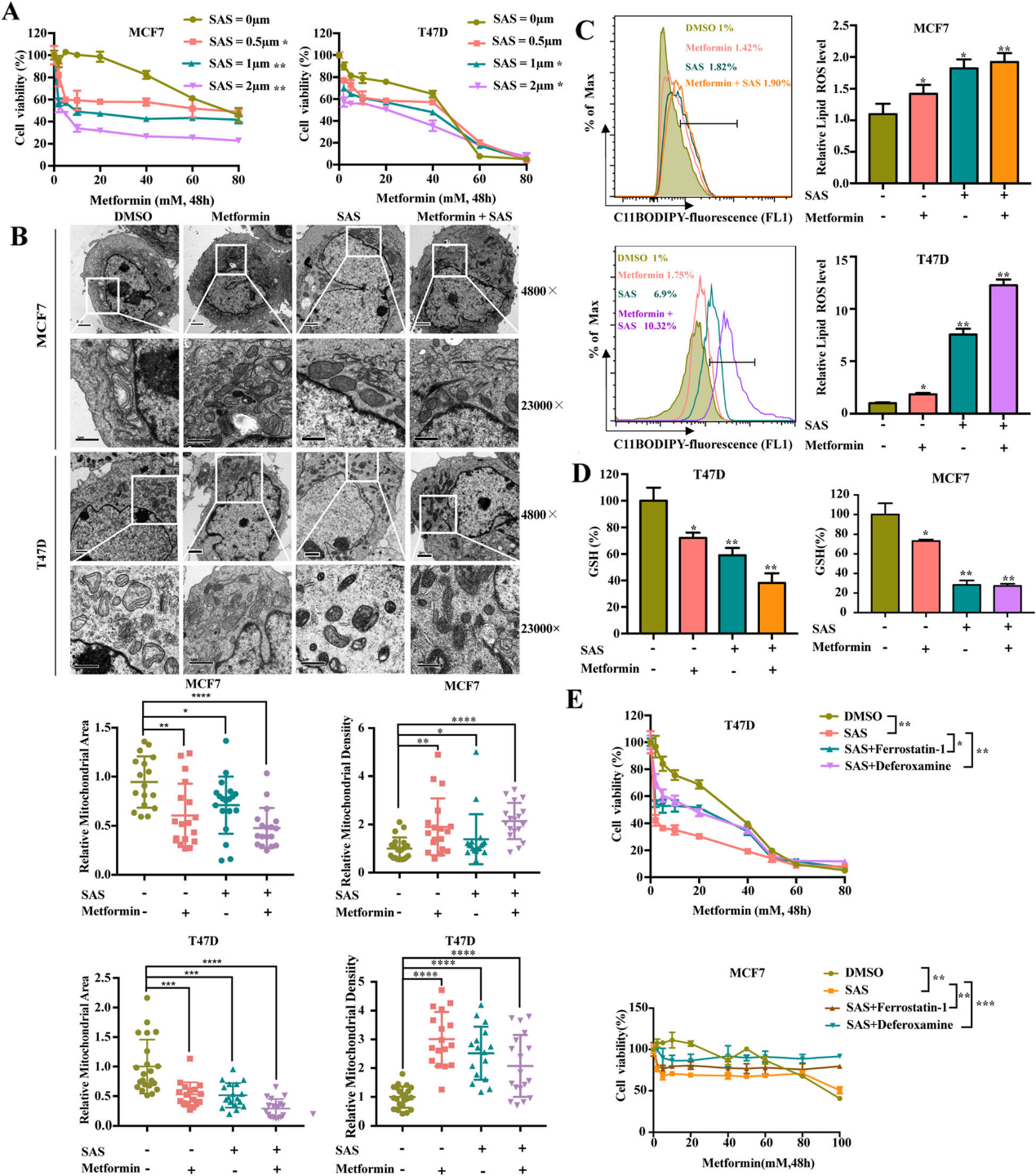
7)SAS与二甲双胍的协同作用能有效抑制乳腺癌细胞
我们用二甲双胍联合SLC7A11抑制剂磺胺吡啶处理细胞。随后,我们采用CCK8法检测细胞活力,以确定二甲双胍与磺胺吡啶联合使用的协同效应。结果表明,磺胺吡啶和二甲双胍联合作用可以协同抑制乳腺癌的增殖,特别是MDAMB231细胞(图7A)。此外,磺胺吡啶与二甲双胍联合使用诱导线粒体体积减小,膜密度增加(图7B)。脂质过氧化检测和谷胱甘肽检测显示,二甲双胍和磺胺吡啶联合使用显著提高脂质ROS水平,抑制谷胱甘肽的生成(图7C, D)。DFO和Fer-1有效地减弱了二甲双胍和磺胺吡啶联合用药的杀伤效果(图7E)。我们的数据表明,磺胺吡啶和二甲双胍联合使用可协同诱导脂质过氧化和铁死亡,从而抑制乳腺癌的增殖。
8)磺胺吡啶可协同增强二甲双胍的体内抗癌活性
我们研究了磺胺吡啶是否能增强二甲双胍的体内抗癌活性。与DMSO组相比,二甲双胍和磺胺吡啶明显抑制肿瘤生长(图8A和B)。在四组中,二甲双胍和磺胺吡啶联合抑制肿瘤生长的效果最高。此外,二甲双胍和磺胺吡啶处理的小鼠表现出较低的UFM1和SLC7A11表达和较高的4-HNE表达(图8C)。这些数据表明,磺胺吡啶可使癌细胞对二甲双胍诱导的脂质过氧化和铁死亡敏感,从而在体内抑制肿瘤。

结论:这项研究是第一次建立二甲双胍和铁死亡之间的联系。我们的数据提示了二甲双胍的一种新的抗癌机制,即二甲双胍通过抑制SLC7A11的UFMylation而诱导铁死亡。