






脂肪干细胞生态位重组结直肠癌干细胞转移机制

肥胖是癌症恶化的一个重要风险因素,与肥胖相关的癌症是导致死亡的主要原因之一。然而,在受肥胖影响的患者中,赋予癌细胞转移特性的分子机制仍未被探索。在此,有作者通过重编程结肠癌(CRC)的4个共识分子亚型(CMS),研究了内脏脂肪组织(VAT)在CRC细胞间质表型调控中的旁分泌效应,证明了微环境细胞因子是预测肥胖相关癌症患者肿瘤行为的重要预后分子。该研究于2021年8月发表在《NATURE COMMUNICATIONS》,IF:12.121。
技术路线:

主要研究结果:
1. 内脏脂肪基质细胞促进CR-CSphCs的致瘤性和转移活性
为了证实肥胖对癌细胞生物学行为的临床影响,作者首先评估了脂肪组织在CRC进展中的潜在作用,以及健康体重(18.50 < BMI < 25)或受肥胖影响(BMI > 30)。对大肠癌患者队列的荟萃分析显示,肥胖与生存概率呈负相关(图1a)。这种相关性也被一项大型队列CRC患者的无进展生存(PFS)分析所证实,该分析认为BMI是独立于分期和治疗的阴性预后因素(图1b)。对患有肥胖症的大肠癌患者肿瘤切片的免疫组化分析强调,脂联素高表达的脂肪细胞位于肿瘤浸润前沿,分布在表达CDX2的肿瘤细胞之间,覆盖了整个肿瘤肿块的28% (图1 c)。同样,肥胖患者的大肠癌肝转移在转移灶边缘,瘤周出芽附近显示出明显的脂肪细胞。在瘦肉型患者的原发性和转移性结直肠癌组织中未观察到这种现象(图1 c)。瘦CRC患者的肿瘤标本显示有表达内皮表型CD34 + / CD31 + /CD45的细胞(图1d)。体外限制稀释试验表明,用V-ASC释放因子处理可提高大量和富集的Wntlow-CRCSPCs的球形起始细胞频率,重现Wnthigh细胞的克隆形成潜能(图1e-g)。这一现象与Wntinactive (GFP-)细胞向Wntactive (GFP +)细胞的转化相平行,证实了Wnt信号通路在CRC干细胞相关重编程中的关键作用(图1h)。V-ASCs与CR-CSphCs共注入小鼠肾包膜下后,致癌细胞的频率增加了 (图1 i)。与更明显的侵袭潜能相一致,在V-ASCs存在的体内限制稀释试验显示,即使在少量细胞被移植时,CMS2 CR-CSphCs也能产生转移到肝脏和肺部的病变(图1j)。这些数据表明ASCs和CRC细胞之间存在支持癌症进展的串扰。
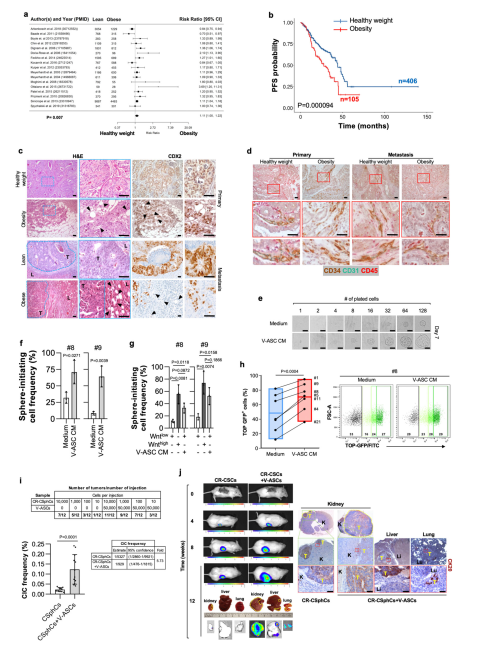
图1浸润肿瘤的VAT提高了CSphCs的转移潜能
2. IL-6和HGF增加了CD44v6 + CR-CSCs的数量,产生NGF,有利于内脏ASCs的迁移能力和内皮转分化
为了确定VAT促进转移的关键分子,接下来研究V-ASCs释放的细胞因子是否会影响CR-CSphCs的转移特性。与SASCs、CR-CSphCs和原代脂肪组织细胞(AT)相比,从肥胖CRC患者中分离出来的V-ASCs产生的IL-6和HGF更丰富 (图2a)。IL-6和/或HGF的存在增强了CR-CSphCs的增殖能力和集落形成潜能(图2b, c),同时获得侵袭表型以及干细胞和转移相关基因的表达,如WNT5A、WNT5B、WNT7A、MMP2、MMP9、TWIST、NODAL、SDF1和ZEB2 (图2d、e)。IL-6和HGF增加了ZEB2蛋白的表达,与其mRNA水平的上调一致(图2f)。此外,阻断IL-6和/或HGF可消除VASCS释放蛋白诱导的CR-CSphCs的细胞增殖和迁移活性(图2g, h)。因此,释放到V-ASC CM中的V-ASC衍生的IL-6和HGF显著促进CD44v6的表达(图2i),CD44v6是一种识别具有强大转移潜能的结直肠癌细胞的标记物,甚至诱导CD44v6球形细胞获得CD44v6表达(图2j)。由于ASCs以CD271的表达为特征,作者评估了其配体(由CSCs产生)在ASCs募集中的作用。与其他CD271配体家族成员BDNF和NTF4(图2k)相比,CD44v6+细胞表达高水平的神经生长因子(NGF)和NTF3 mRNA,并分泌NGF和NT-3,而这些神经营养素很少被CD44v6-细胞释放,除非暴露于V-ASC的CM(图2l)。CD44v6+细胞释放的NGF和NT-3促进ASCs募集,与NGF中和抗体的较弱作用相比,CD271中和抗体可完全阻止ASCs募集(图2m)。此外,暴露于V-ASC CM的CR-CSPCs也能够吸引ASC(图2m,n)。因此,这些数据表明,ASC衍生的IL-6和HGF可以将CD44v6-初始细胞重新编程为CD44v6+细胞,从而通过分泌NGF和在肿瘤肿块内招募ASC来增加其转移潜能。
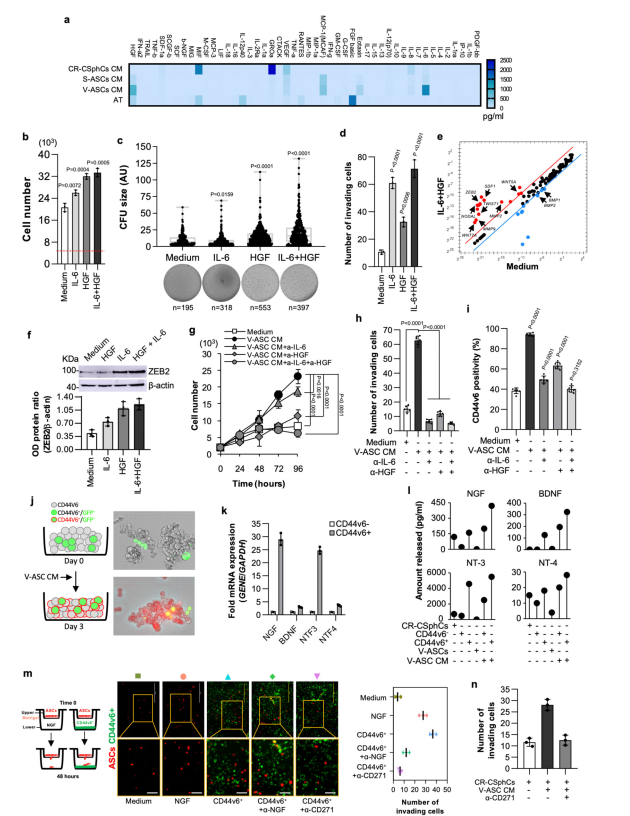
图2脂肪源性因子扩大了分泌NGF的CD44v6 +细胞片段,增强了ASCs的迁移能力
CD44v6 +细胞表达并产生高水平的VEGF,增加了ASCs的增殖率(图3a, b)。另外,大多数CD271 + ASCs表达VEGFR(图3c),提示CD44v6 +细胞来源的VEGF可能触发ASCs的血管生成信号。CD44v6+细胞的旁分泌活性诱导富集的CD34+/CD31 ASC向表达CD31的内皮样细胞转分化(图3d)。此外,暴露于CD44v6+细胞释放的CM导致HUVEC内皮细胞形成血管小管,类似于VEGF治疗后形成的血管小管(图3e和补充图3d)。在共注射CR-CSphCs和V-ASCs生成的结CRC异种移植物中,与更明显的血管密度相关的CD44v6 +细胞优先定位在血管前部(图3f),这些发现表明CSC腔室参与了新血管生成和已有毛细血管的新生萌芽。
3. 内脏释放蛋白诱导CR-CSphCs的EMT
CR-CSphCs暴露于V-ASC CM中,证实了释放蛋白在触发转移途径中的重要作用,将转录组结构从上皮/CMS2模式转变为类似于间质CMS4的表型(图3g)。同样,对单独使用V-ASC CM或培养基处理的CR-CSphCs进行的RNA-Seq转录组分析显示,有10个DEGs与CMS4特征相关(图3h)。IL-6和HGF靶向消除VAT诱导的转移活性,恢复CMS2 CR-CSphCs的非转移表型(图3i)。这些结果表明V-ASCs旁分泌活性驱动CRC球细胞向间充质样表型转变,赋予它们转移潜能。
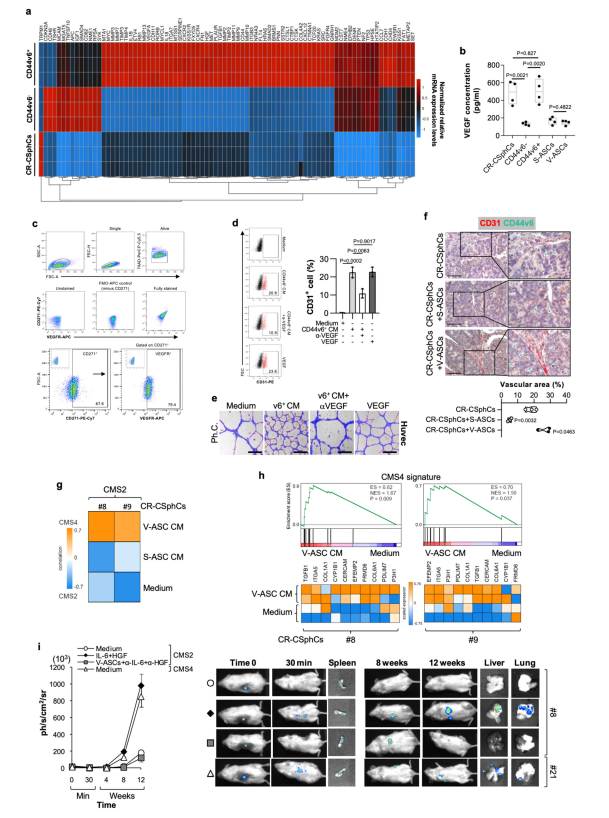
图3 VEGF诱导ASCs内皮分化,激活CRC球细胞EMT程序
4. VAT通过调控ZEB2表达来管理EMT
利用分子特征数据库(MSigDB)进行的基因集富集分析(GSEA)显示,与代谢途径相关的基因呈负富集,与EMT程序相关的基因呈正富集(图4a)。上调E-box结合同源盒(ZEB)转录因子ZEB1和ZEB2,与释放的蛋白重编程CMS2 CR-CSphCs向转移前表型的能力一致(图4b)。在V-ASC CM处理的CMS4和CMS2 CR-CSphCs中,有15个基因表达上调,6个基因表达下调(图4c)。在V-ASCs细胞因子的诱导下,ZEB1和ZEB2的mRNA和蛋白水平均上调(图4d, e),有报道称ZEB1和ZEB2可调节miR-200家族成员的表达并调节CRC中的EMT。对miRNA表达谱分析显示,V-ASC CM处理后,CMS2 CR-CSphCs中miRNA-200家族成员,特别是miR-200a的表达下调,显示出与CMS4 CR-CSphCs相似的miRNA水平(图4f)。在使用V-ASCs CM处理的CR-CSphCs中,对大多数差异表达mirna网络及其靶标的分析表明,ASC释放的蛋白调控了大量聚集在miR-200a/ZEB成员轴上参与EMT过程的基因(图4g)。在V-ASC CM作用下,CMS2细胞中ZEB2蛋白的表达水平与CMS4细胞中相似(图4h)。此外,外源表达ZEB2可促进vimentin和CD44v6的表达,并增强肿瘤向远处器官的扩散(图4i、j),表明ZEB2可能作为具有转移特性的CRC细胞的功能标记物。总之,这些结果表明ZEB2是具有转移潜能的CRC的功能性生物标志物。
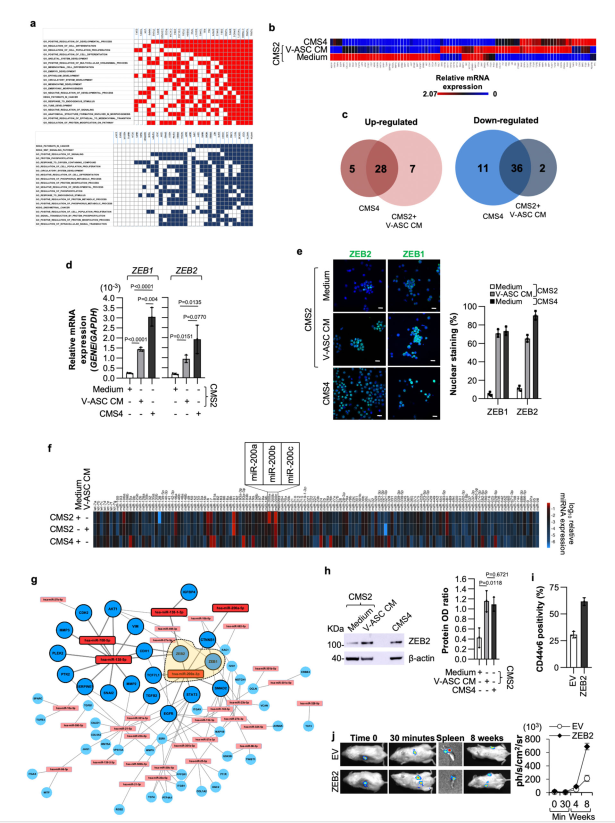
图4 V-ASCs增强ZEB2的表达,维持CR-CSPCs的转移活性
5. IL-6/HGF阻断降低VAT诱导的转移形成
GSEA显示,V-ASC释放因子促进了CR-CSPCs中STAT3激活途径相关基因的富集(图5a)。免疫印迹分析显示,V-ASC CM和IL-6/ HGF均通过增强酪氨酸和丝氨酸残基中STAT3的磷酸化来激活CMS2细胞中的STAT3通路(图5b)。根据结肠肿瘤切片免疫组化分析,STAT3的激活主要位于肥胖CRC患者的脂肪组织附近,细胞核染色突出。而在瘦骨嶙峋的患者中,与肿瘤间质接触的癌细胞表现出核p-STAT3的微弱存在(图5c)。STAT3磷酸化抑制剂C188-9降低了由IL-6和HGF共同维持的STAT3的激活(图5d)。此外,在IL-6和HGF的存在下,C188-9显著降低了细胞增殖率和克隆活性,并恢复了CR-CSphCs中miR-200a和ZEB2的表达水平(图5e h)。同样,暴露于IL-6和HGF的CMS2 CR-CSphCs,其细胞形态发生了变化,获得了与拉长形状相关的独特极化,而暴露于C188-9则减弱了这种极化(图5i)。这些结果表明,脂肪微环境细胞因子通过STAT3/miR-200a/ZEB1/2轴驱动上皮相关CMS2 CR-CSphCs获得间充质表型。

图5 IL-6和HGF通过激活STAT3诱导间质表型
托西珠单抗和克唑替尼的联合治疗能够恢复V-ASC CM对CR-CSphCs增殖和集落形成能力的影响(图6a, b)。此外,这种双靶点治疗阻碍了v - asc诱导的miR- 200a下调,从而降低了ZEB1和ZEB2的表达,并降低了CMS2 CR-CSphCs的获得性侵袭行为(图6c-e)。为了确定托西珠单抗联合克唑替尼是否可以用于辅助治疗,防止内脏肥胖诱导的转移形成,将CMS2 crcsphc和V-ASCs共注射到小鼠脾脏中产生转移性小鼠替身。脾切除术后5天,小鼠每周治疗3次,持续3周(图6f)。,这种治疗方案显著降低了V-ASC刺激的CMS2细胞的转移性植入频率,即使是在治疗暂停8周后(图6g)。此外,对112例CMS2 CRC患者的队列分析显示,ZEB2表达与RFS概率显著负相关(图6h, i),提示ZEB2可能是一个推测的预后因素,可能与肥胖影响的癌症患者相关。

图6 IL-6和HGF靶向抑制CR-CSphCs VAT诱导的转移能力
主要结论:
作者发现通过抑制STAT3 (C188-9)、IL-6R(托西利珠单抗)或c-MET(克唑替尼)靶向JAK/STAT通路的激活,会损害CMS2 CR-CSphCs的VAT驱动转移促进活性(图6i)。因此,由于脂肪释放因子通过确定慢性炎症状态和增加远处转移形成的风险来促进CRC微环境的形成,这些药物的临床可用性可以被考虑作为肥胖相关癌症患者的辅助治疗策略。