






巨噬细胞已经过时了吗?非也,看过来学习新思路
肿瘤相关巨噬细胞(TAMs)支持肿瘤进展在响应肿瘤和基质细胞构成的微环境中扮演着促肿瘤表型作用。但是肿瘤细胞指示TAM行为的潜在机制仍然未知。该文发现肿瘤-细胞-来源的葡萄糖神经酰胺刺激的非传统性内质网压力响应通过诱导巨噬细胞内质网膜的脂质组分重组和饱和,这诱导了IRE1-介导的XBP1剪切和STAT3激活。这种肿瘤-细胞-产生的脂质通过内质网应激反应同时协调巨噬细胞极化和肿瘤存活的作用,可能是维持宿主抗肿瘤免疫的治疗靶点。本文于2021年10月发表在《Nature Immunology》IF: 25.606杂志上。
技术路线:

主要实验结果:
为了了解TAM的脂质代谢,作者在YUMM1.7黑素瘤小鼠移植模型和基因工程小鼠黑素瘤模型(简称Braf/Pten小鼠)中,我们通过bodypy和FilipinIII染色和bodypy C12(一种荧光脂类类似物)摄取来评估肿瘤和脾脏中巨噬细胞的脂质含量和摄取。在这两种模型中,与脾脏巨噬细胞相比,TAMs增加了中性脂质(BODIPY染色)和胆固醇(Filipin染色)含量和摄取(图1a,b)。此外,脂滴只出现在TAM的细胞质中,而不出现在脾脏巨噬细胞中(图1c)。致瘤功能的标记酶ARG1阳性的巨噬细胞的脂质含量高于ARG1阴性的TAMs(图1d),提示TME引起的异常脂质积累可能会扭曲TAMs的致瘤特性。除了脂滴的形成外,我们还发现TAMs的内质网更加扩张和肿胀,这是内质网应激反应的形态学标志(图1e)。以上表明TAMs具有更高的脂质含量和ER应激反应。
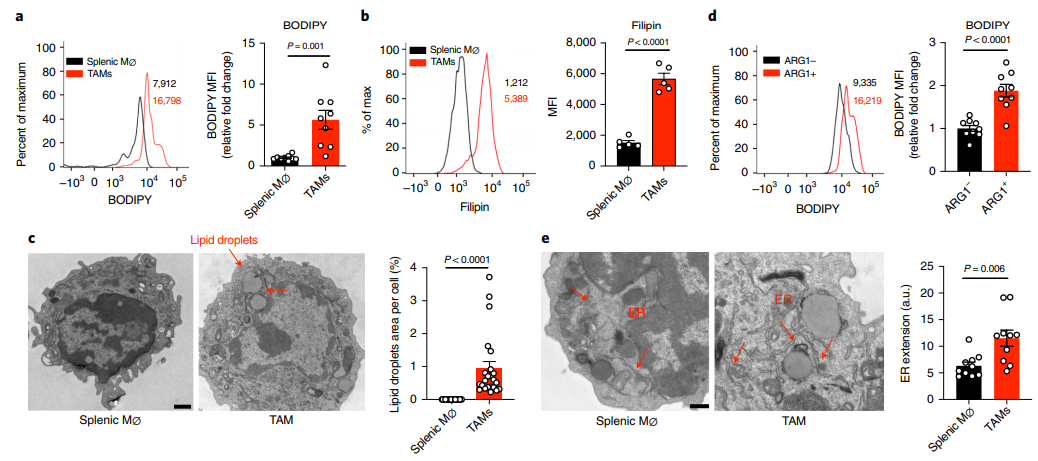
图1 TMA促进致瘤性TAMs的脂质积累
进一步支持了上述结论,研究发现TAMs增加了典型内质网逆境应答基因的mRNA表达,包括Ern1, Bip和剪接Xbp1 (sXBP1),无论是在移植还是诱导的黑色素瘤模型中(图2a),并且,与脾脏巨噬细胞相比,TAMs中sXBP1蛋白的表达率更高(图2b),表明TAMs可能参与内质网应激反应。由于最近的研究表明,脂质代谢失调和内质网应激之间的相互干扰在调节代谢组织的细胞行为方面发挥了重要作用,作者接下来检测了脂质含量sXBP1和ARG1在TAMs中的表达。结果显示sXBP1+ TAMs 具有更高的脂质含量(图2c)和更高组分的致瘤性ARG1+ TAMs(图2d)。通过计算几种人类癌症,包括大肠癌、无慢性阻塞性肺疾病的肺腺癌(LUAD-No COPD)和非小细胞肺癌(NSCLC)和一组小鼠肉瘤的单细胞RNA测序结果中单个TAM的内质网应激反应,作者观察到,与抗肿瘤TAMs相比,不同癌症中的促肿瘤TAMs都表现出更高的内质层应激得分(图2e)。综上所述,内质网应激和脂质代谢失调可能相互协调,从而诱导TAMs的致瘤性特征。
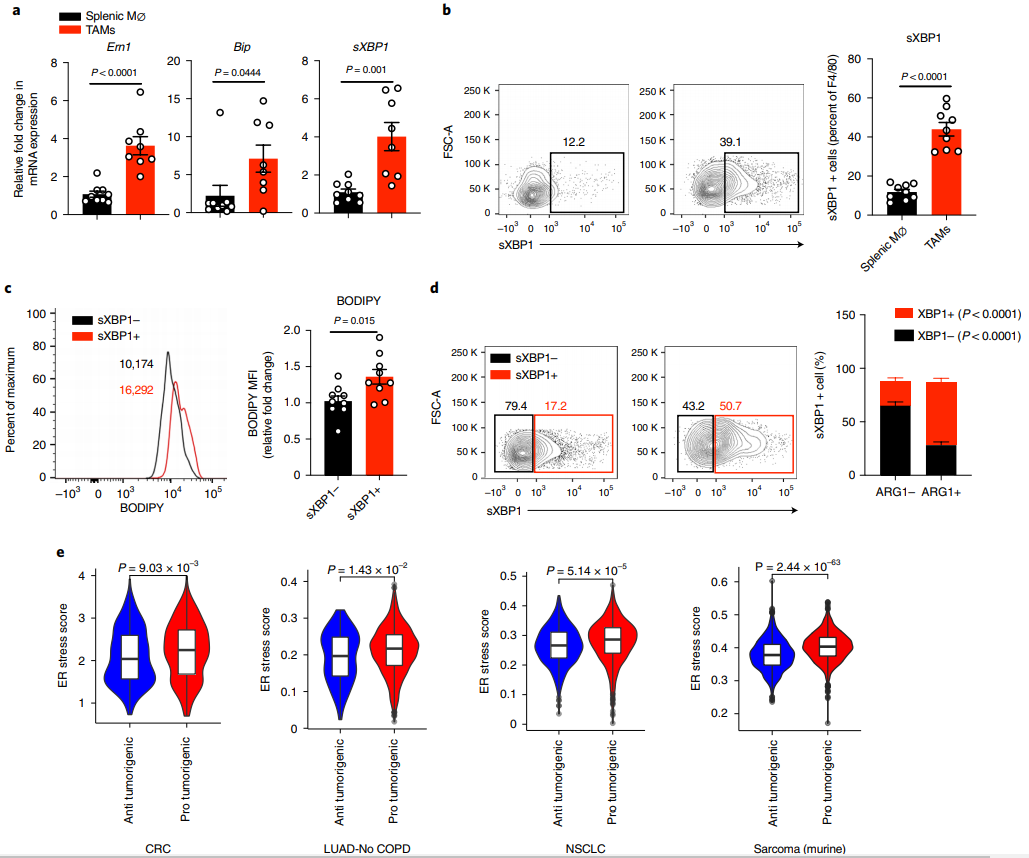
图2 TME激活致瘤性TAMs的IRE1/sXBP1通路
2、IRE1-XBP1增强巨噬细胞肿瘤前极化
为了探究是否肿瘤-细胞-来源因子可以刺激增加巨噬细胞的脂质含量和内质网应激,作用用黑色素瘤YUMM1.7肿瘤-细胞-来源的条件培养基(CM)处理骨髓来源巨噬细胞(BMDM)。结果显示,与对照相比,CM显著促进BMDM的脂质含量和摄取(图3a),相应的,促肿瘤标志物基因的表达也显著增加,如Arg1和Mrc1(图3b)。与naïve巨噬细胞相比,CM处理的BMDM具有更高的抑制CD8+ T细胞增殖的活性
此外,CM处理的BMDM表现出sXBP1的产生,但是PERK的激活和下游靶基因ATF4表达少量增加(图3c)。鉴于内质网应激反应最近被揭示通过调节CD8+ T细胞、树突状细胞和MDSCs的功能来抑制抗肿瘤免疫,作者推测CM介导的IRE1-XBP1信号的激活可能支持了巨噬细胞获得致瘤表型。为了验证这一假设,用STF081030处理CM刺激的BMDM,STF081030是一种抑制IRE1的核糖核酸内切酶活性的抑制剂,可以防止sXBP1的产生,结果发现STF081030有效地抑制了由CM引起的sXBP1和促肿瘤标志物基因的表达(图3d)。此外,STF081030处理有效改善了CM对BMDM的抑制能力(图3e)。
为了证实IRE1在诱导前致瘤极化中的作用,将LysM-Cre Cas9小鼠产生的BMDM分别用含有scramble guide RNAs (gRNAs)或IRE1靶向gRNAs的慢病毒转导,以产生对照BMDM或IRE1缺陷BMDM。与对照组BMDM相比,IRE1缺陷BMDM中IRE1表达较低,研究发现CM诱导的致瘤前极化在IRE1缺陷BMDM中被抑制(图3f),这表明CM诱导的IRE1活性增强了巨噬细胞中致瘤前极化。传统的内质网应力诱导剂,包括tunicamycin和thapsigargin在内,均能诱导内质网应激反应,但不能使BMDM分化为致瘤表型(图3g)。以上表明,与传统内质网应激反应相比,sXBP1控制的特殊信号级联反应是加强巨噬细胞对肿瘤细胞衍生因子反应的免疫抑制活性所必需的。

图3 肿瘤细胞通过IRE1在BMDM中驱动致瘤前极化
3、XBP1重塑TAM表型并支持肿瘤进展
为了进一步阐明XBP1对巨噬细胞致瘤前极化的贡献,将LysM-Cre Cas9小鼠产生的BMDM分别用含有杂合GRNAS或XBP1靶向GRNAS的慢病毒转导,以生成对照BMDM或XBP1缺陷BMDM。与对照组相比,XBP1缺陷BMDM中XBP1总表达和剪接表达均降低。结果发现,与对照组BMDM相比,XBP1缺陷BMDM中CM诱导的致瘤标志物基因表达减少(图4a)。此外,与XBP1野生型小鼠相比,CM对小鼠的免疫抑制能力在XBP1缺失后显著消失,表现为免疫细胞数增多(图4b),并且XBP1缺失小鼠的肿瘤体积和重量也都显著下降(图4c, d),提示XBP1可使巨噬细胞向致瘤前表型倾斜。作者通过检测WT和XBP1cko小鼠的TAMs,发现XBP1缺失导致了mTAMs和iTAMs丰度的变化,以及iTAMs与mTAMs的比例降低(图4e),表明敲除XBP1可以改善TAMs的致瘤性特征。总之,XBP1的表达促进TAMs向致瘤表型激活。

图4 TAMs中XBP1缺失抑制肿瘤生长
4、STAT3信号优化巨噬细胞肿瘤前极化
接下来,探究sXBP1单独表达是否足以促进M2表型。结果显示,STF083010处理显著抑制了对照BMDM细胞中CM诱导的致瘤性标志物基因的表达,但对过表达sXBP1的BMDM没有影响(图5a)。支持了此前的结论,即在CM刺激的巨噬细胞中,需要IRE1介导的sXBP1的产生来增强原致瘤活性。
研究表明,STAT3的激活通过触发TAMs中的组织蛋白酶表达来促进肿瘤进展。作者发现,CM处理显著增加了STAT3的磷酸化(图5b)。推测,STAT3可能也是增强肿瘤-细胞CM的致瘤极性所需要的。研究显示STAT3的基因缺失或STAT3抑制剂Stattic的处理会降低Mrc1基因的表达,但不会影响Arg1的表达(图5c),这表明STAT3的激活可能在一定程度上参与了CM处理BMDM的抑制活性。接下来,检测了肿瘤-细胞是否可以通过IL-6和IL-10的产生激活BMDM中的STAT3。结果发现抗体IL - 6和IL - 10无法抑制CM-Induced STAT3磷酸化(图5d)和致瘤性标记基因的表达(图5e),而该剂量的抗体能够废除导IL - 6和IL – 10诱导的STAT3磷酸化(图5d)。些结果表明,肿瘤-细胞-来源因子激活STAT3,以IL-6/IL-10不依赖的方式促进巨噬细胞的致瘤前极化。
IRE1已被证明通过与STAT3形成蛋白复合物来支持肝细胞中STAT3的激活。通过使用邻近连接试验,发现CM促进了BMDM中的IRE1-STAT3相互作用(图5f)。然后,检测STAT3磷酸化是否需要IRE1,发现与对照组BMDM相比,肿瘤细胞CM诱导的STAT3磷酸化在IRE1缺陷的BMDM中被抑制(图5g)。总之,以上结果表明CM触发的IRE1信号通过同时刺激STAT3磷酸化和sXBP1的产生,导致巨噬细胞的致瘤极化。

图5激活IRE1-STAT3信号支持CM诱导极化
5、Mincle感知葡萄糖神经酰胺使巨噬细胞活化
由于脂质代谢异常被认为是诱发内质网应激反应的原因,作者推测,肿瘤细胞产生的脂质可能是诱导巨噬细胞致瘤前特征的原因。为了验证这一点,使用脂质去除剂从YUMM1.7黑色素瘤细胞CM中去除脂质,包括胆固醇和脂肪酸。结果发现CM脂质去除后,CM引发事件的能力也都消失了,包括sXBP1产生、STAT3磷酸化和致瘤标志物基因的表达(图6a-c)。检测CM和脂质去除CM的转录组,发现巨噬细胞中大多数负责致瘤活性的基因以脂依赖的方式在肿瘤细胞CM中上调(图6d)。肿瘤细胞产生的脂质可能控制内质网应激参与的巨噬细胞致瘤性极化。
通过检查转录组分析,作者重点关注参与脂质基因识别和绑定的,并在CM组显著上调的基因,发现基因——巨噬细胞诱导Ca2+依赖外源凝集素受体(Mincle),也称为C型凝集素域家族4成员E (Clec4e),显著响应CM处理(图6e)。进一步研究发现,CM对BMDM的免疫抑制,上调致瘤性标志基因表达,和STAT3磷酸化的作用,都能被anti-Mincle抗体所反转(图6f-h)。总之,这些结果表明,Mincle介导的脂质识别是CM诱导巨噬细胞致瘤前极化所需要的。
由于β-葡萄糖神经酰胺和胆固醇硫酸盐是Mincle已知的内源性配体,所以作者研究了YUMM1.7细胞中β-葡萄糖神经酰胺的产生是否是BMDM中驱动CM诱导的致瘤前极化所需要的。构建针对UDP -葡萄糖神经酰胺葡萄糖基转移酶(UGCG)的shRNA,UGCG是β-葡萄糖神经酰胺生成的代谢酶,以减少YUMM1.7黑色素瘤细胞中UGCG表达和β-葡萄糖神经酰胺生成(图6i)。结果显示,和对照相比,UGCG缺失的CM对巨噬细胞致瘤性诱导的能力显著下降,表现为UGCG缺失组的致瘤性标志物基因表达下降,STAT3磷酸化下降,肿瘤的体积和重量下降,iTAM/mTAM比值下降(图6j-n)。总之,这些结果表明,肿瘤细胞产生的β-葡萄糖神经酰胺以Mincle依赖的方式触发内质网应激反应,释放TAMs的致瘤活性。
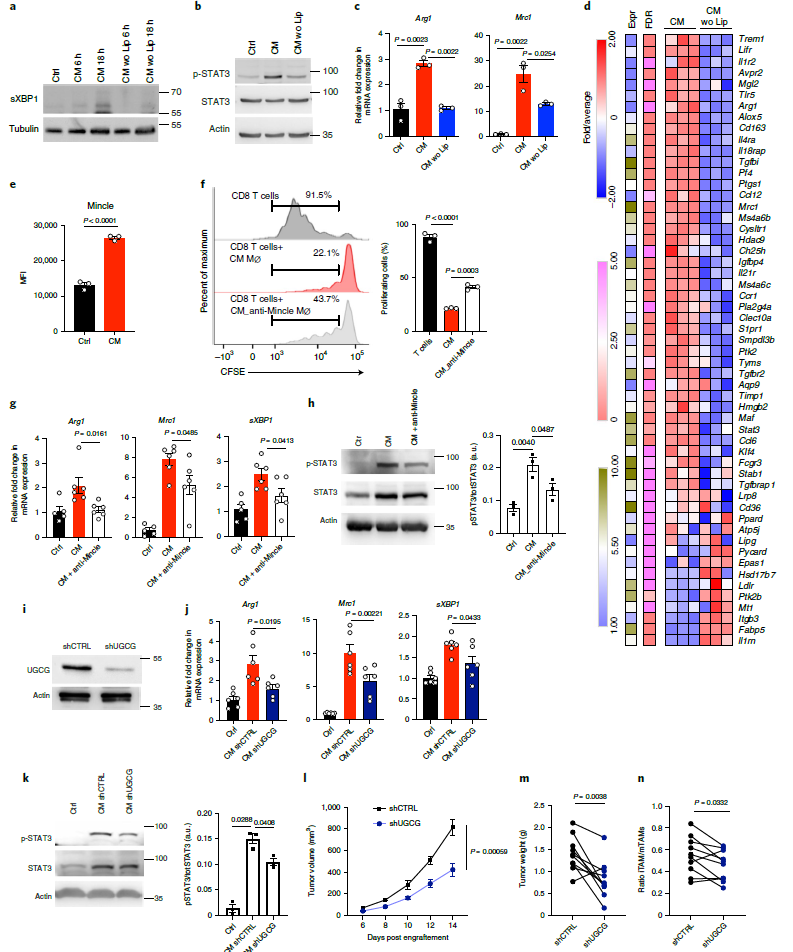
图6 Mincle依赖的葡萄糖神经酰胺感知途径使巨噬细胞活化
6、内质网膜脂质成分紊乱激活sXBP1
鉴于文献表明Mincle可以抑制胆固醇外排,以及TAM可以积累更多的胆固醇,所以作者推测CM可能是促进细胞内胆固醇积累通过刺激胆固醇合成。Fillipin染色结果表明CM可以促进细胞内胆固醇含量,但是他汀处理阻断胆固醇合成以及anti-Mincle抗体处理都可以有效的阻碍CM诱导的胆固醇积累(图7a)。此外,他汀类药物阻断胆固醇合成也阻止了CM诱导的sXBP1产生和致瘤性标志物基因在BMDM中的表达,但是他汀类药物和anti-Mincle抗体协同作用不会进一步放大该效应(图7b)。IRE1含有一个跨膜结构域,可以感知脂质失衡并诱导其二聚和激活,增加胆固醇积累可能会使内质网膜的脂质成分重组为较低的磷脂酰胆碱与磷脂酰乙醇胺比值(PC/PE比值)和较低的多不饱和脂肪酸(PUFA),它会降低内质网的膜流动性,这就是胆固醇感应机制紊乱的结果。因此,作者推测肿瘤-细胞CM通过重组内质网膜上的脂质组分来触发IRE1/XBP1的激活。内质网膜脂质谱显示,与对照组相比,CM处理的BMDM的PC/PE比值显著降低,并观察到PUFA的比值显著下降(图7d, e)。随后,作者通过过表达LPCAT3(一种PC合成酶)试图挽救内质网膜脂质组分。结果发现,过表达LPCAT3显著增加了CM刺激的内质网膜的PC/PE比值和不饱和PC的丰度(图7f-h),还观察到LPCAT3过表达减少了CM处理的BMDM内质网膜的延伸(图7i),以及阻断CM诱导的sXBP1产生和致瘤性基因的表达(图7j-l)。综上所述,这些数据表明,肿瘤诱导的内质网膜脂质成分重组对启动内质网应激介导的致瘤极化至关重要。

图7 CM引起内质网膜脂质重组和饱和
7、LXR激动剂通过LPCAT3控制巨噬细胞的肿瘤负荷
前人研究表明LPCAT3的表达受到LXR的调控,所以作者试图用LXR激动剂GW3965诱导LPCAT3的表达,并研究这种处理是否可以用来定制TAMs的功能。GW3965在CM处理的BMDM中诱导LPCAT3(图8a),并降低sXBP1产生,STAT3磷酸化,致瘤性基因的表达,部分缓解CM对CD8+T细胞的抑制性(图8b-e)。此外,GW3965药物处理可以减少肿瘤的体积和重量,表明GW3965可以抑制肿瘤进展,这和此前报道一致(图8f-g)。为了进一步鉴定抗肿瘤反应是否依赖于巨噬细胞中LPCAT3的表达,构建了LPCAT3敲除鼠。结果显示,在WT小鼠中,GW3965处理能显著抑制肿瘤进展,而LPCAT3敲除后,这种抑制作用消失了(图8h-l)。总的来说,与之前从xbp1缺陷小鼠获得的数据一致,内质网应激反应,特别是sXBP1的表达,对于TAMs的生存和致瘤极化是必需的,并且LXR介导的巨噬细胞LPCAT3诱导可以被利用来唤醒抗肿瘤反应。
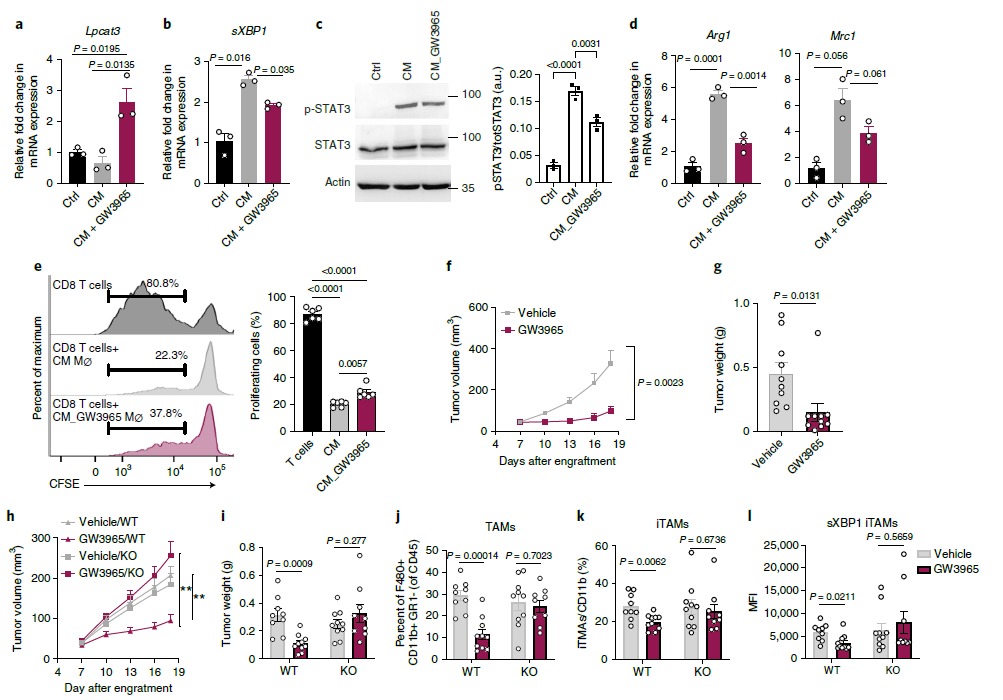
图8 LXR激动剂以LPCAT3依赖的方式减少肿瘤负担并阻碍TAM的生存