






Rab2A通过稳定PPARγ调控AMPK-TBC1D1轴下游非酒精性脂肪性肝病的进展
非酒精性脂肪肝(NAFLD)的发病率约占全世界人口的四分之一,持续的营养过剩是其主要原因之一。然而,其潜在的分子基础尚未完全阐明,也没有专门的药物被批准用于这种疾病。目前,由作者发现了一个调控机制,揭示了Rab2A在NAFLD进展中基于能量状态和PPARγ的新功能。该研究于2022年1月发表在《PLoS biology》,IF:8.029。
技术路线:

主要研究结果:
1.阻断AMPK-TBC1D1轴导致老年小鼠肝脏脂质沉积
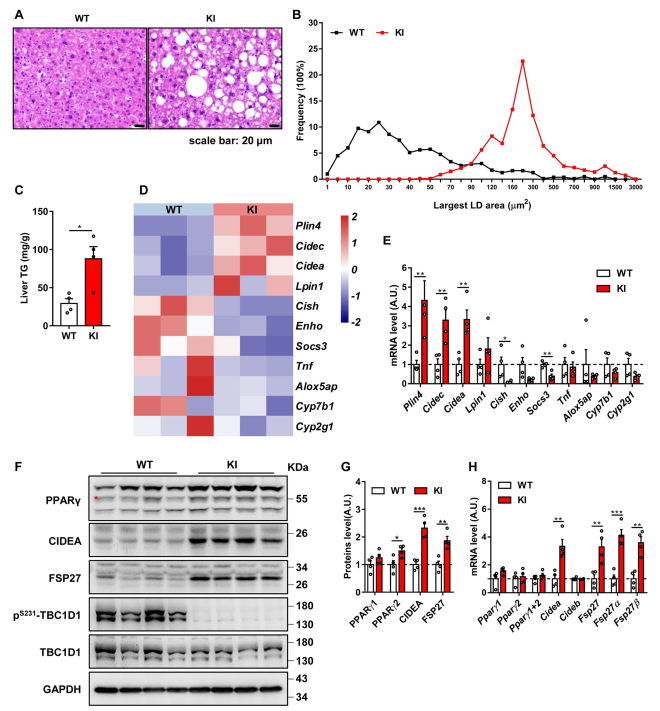
作者在4 - 6月龄、12月龄和18月龄的TBC1D1-KI小鼠中检测了NAFLD相关表型:12月龄和18月龄TBC1D1-KI小鼠肝脏切片大脂滴积累显著增加(图1A和1B),同时TG水平严重升高(图1C)。上述数据表明,阻断AMPK-TBC1D1轴导致老年小鼠肝脂积累。然后对18个月大的小鼠肝脏进行RNA测序,热图分析脂质沉积类基因Cidea、Cidec和Plin4表达增加,它们是PPARγ的靶基因(图1D),q-PCR结果也证实了这些发现(图1E)。此外,还观察到12月龄和18月龄TBC1D1-KI小鼠PPARγ蛋白水平显著升高,而PPARγ mRNA水平无明显差异(图1F-1H),这些效应导致PPARγ活性升高。综上所示,阻断AMPK-TBC1D1轴可能通过激活PPARγ信号通路导致老年小鼠发生NAFLD。
2. AMPK-TBC1D1轴调控PPARγ的蛋白稳定性和功能
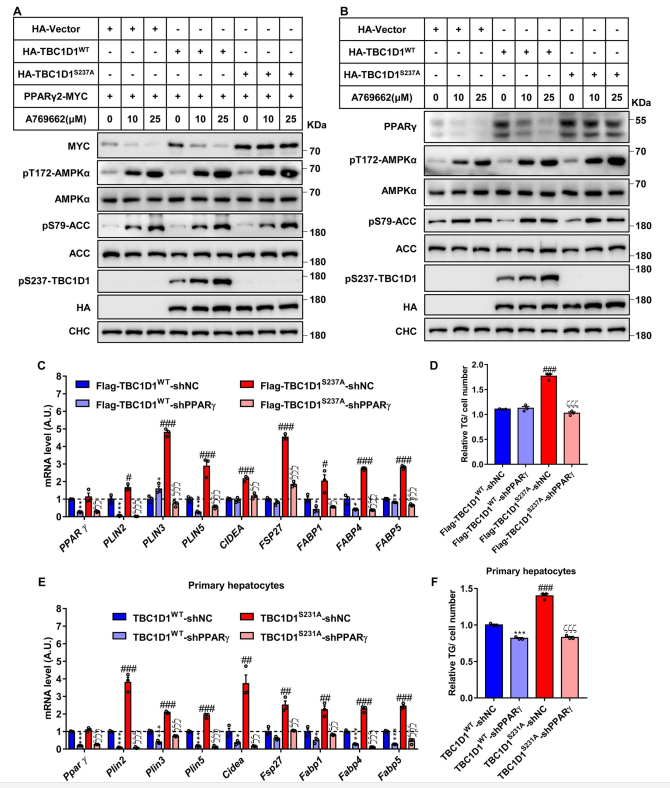
AMPK激活剂A769662处理以剂量依赖的方式增加AMPK及底物乙酰COA羧化酶(ACC)的磷酸化,并且AMPK的激活状态在空载体或WT TBC1D1或TBC1D1S237A突变体的细胞中是相似的(图2A和2B)。A769662激活AMPK后,外源性PPARγ2(图2A)和内源性PPARγ(图2B)逐渐下降,这与A769662的剂量有关。WT TBC1D1的表达同时提高了外源性PPARγ2和内源性PPARγ的表达,而在A769662处理下AMPK激活后,外源性PPARγ2和内源性PPARγ的表达仍然降低(图2A和2B)。在TBC1D1S237A突变体细胞中,A769662诱导的外源性PPARγ2和内源性PPARγ的减少被阻止(图2A和2B)。
接下来研究了TBC1D1 KI突变诱导的脂质储存基因的表达是否确实是由PPARγ介导的。作者采用了两种细胞模型,即表达TBC1D1WT或TBC1D1S237A蛋白的HepG2细胞和来自WT和TBC1D1S231A-KI小鼠的原代肝细胞。TBC1D1S237A突变蛋白的表达显著增加了PPARγ靶基因PLIN3、PLIN5、CIDEA、FSP27/CIDEC、FABP1、FABP4和FABP5在HepG2细胞中的表达(图2C)和细胞中TG含量的升高(图2D)。同样,PPARγ基因的敲除挽救了脂质储存基因如Plin3、Plin5、Cidea、Fsp27/Cidec、Fabp1、Fabp4和Fabp5的表达(图2E),并阻止了TG在TBC1D1S231A-KI小鼠原代肝细胞中的积累(图2F)。总之,这些数据确定了AMPK-TBC1D1轴在调节PPARγ蛋白中的因果作用,并且AMPK-TBC1D1连接的破坏增加PPARγ蛋白,通过提高肝细胞中脂质储存基因的表达促进TG积累。
3. Rab2A作为AMPK-TBC1D1轴的下游蛋白,调控PPARγ的蛋白水平
体外结合实验也证实了TBC1D1和PPARγ2之间的直接相互作用(图3A)。而TBC1D1S237A突变蛋白仍然具有与PPARγ相互作用的能力,其方式与WT TBC1D1类似(图3A),表明TBC1D1- s237磷酸化并不影响TBC1D1与PPARγ的相互作用。GAP非活性TBC1D1R854K突变体的过度表达增加了PPARγ2蛋白,其程度类似于TBC1D1S237A突变体蛋白(图3B)。内源性PPARγ在Rab2A过表达细胞中升高(图3C),而在Rab2A敲低表达细胞中降低(图3D)。在HepG2细胞中,TBC1D1S237A蛋白的过度表达(而非WT TBC1D1)显著增加了Rab2A的GTP结合形式(图3E)。此外,在TBC1D1-KI小鼠(18个月大)的肝脏中,Rab2A的GTP结合形式显著增加,而Rab2A的总水平不变(图3F和3G)。这些数据表明,TBC1D1-S231A突变导致细胞中Rab2A的激活。TBC1D1 WT或S237A蛋白诱导的外源性PPARγ2(图3H)的增加通过shRNA敲除Rab2A而减弱。在HepG2细胞中,Rab2A的敲除阻止了TBC1D1S237A突变蛋白诱导的脂质储存基因,包括PLIN3、CIDEA、FSP27/CIDEC、FABP1和FABP5(图3I),并阻断了TG的积累(图3J)。同样,Rab2A基因的下调也恢复了脂质储存基因Plin2、Plin3、Plin5、Cidea、Fsp27/Cidec、Fabp1、Fabp4和Fabp5的表达(图3K),并阻止了TG在TBC1D1S231A-KI小鼠原代肝细胞中的积累(图3L)。这些数据表明,Rab2A在TBC1D1下游发挥基因作用,调控PPARγ在肝脏脂质储存中的作用。

4.Rab2A的GTP结合形式结合并抑制PPARγ的蛋白酶体降解
在共免疫沉淀试验中,在Flag-Rab2A的免疫沉淀中发现内源性PPARγ(图4A)。在体外GST-pulldown实验中,GST-PPARγ2也能与Flag-Rab2A结合(图4B)。此外,影像学研究显示,部分PPARγ2-mCherry存在于细胞质中,并与主要存在于细胞质中的EGFP-Rab2A共定位(图4C)。Rab2A的GTP结合形式显著提高了内源性PPARγ(图4D)的蛋白水平。此外,在共免疫沉淀试验中,Rab2A的GTP结合形式与PPARγ2-MYC相互作用(图4E)。在共免疫沉淀试验中,AF-2结构域的缺失阻止了PPARγ2与Rab2A的结合(图4F)。这些数据表明,Rab2A的GTP结合活性形式可能与PPARγ的AF-2结构域相互作用,从而提高PPARγ的蛋白质水平。用蛋白酶体抑制剂MG132和ALLN处理,都能以浓度和时间依赖的方式导致PPARγ2-MYC的积累(图4G-4N)。过表达Rab2A显著减弱了这些蛋白酶体抑制剂对PPARγ2-MYC积累速率的影响(图4G-4N)。综上所述,这些数据表明Rab2A的GTP结合形式与PPARγ的AF-2结构域结合,并抑制PPARγ的蛋白酶体降解。

5.营养状态调控AMPK-TBC1D1-Rab2A轴的活性,进而调控PPARγ的蛋白水平
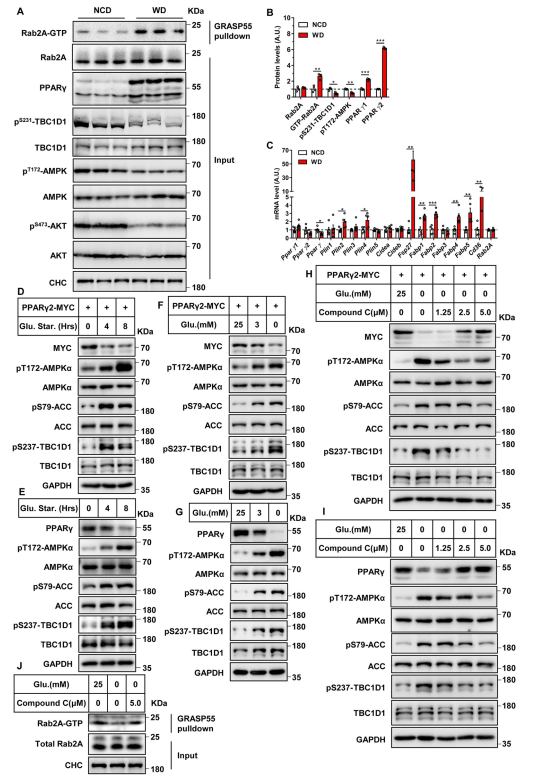
DIO小鼠中的过度营养会使AMPK失活,从而导致肝脏中TBC1D1的磷酸化降低(图5A和5B)。Rab2A在DIO小鼠的肝脏中成为GTP负载的活性形式(图5A和5B)。DIO小鼠肝脏中Pparγ1和Pparγ2 mRNA水平均未发生变化(图5C),而蛋白水平显著升高(图5A和5B)。此外,DIO小鼠肝脏PPARγ靶基因也显著增加(图5C)。这些数据表明,AMPK-TBC1D1-Rab2A轴对营养过剩做出反应,以增加小鼠肝脏中的PPARγ蛋白。在HEK293T和HepG2细胞中,葡萄糖饥饿激活了AMPK通路,AMPK和ACC的磷酸化增加,使TBC1D1在237丝氨酸位点磷酸化(图5D-5G)。葡萄糖饥饿降低了HEK293T细胞外源性PPARγ2- MYC的蛋白水平(图5D和5F),也降低了HepG2细胞内源性PPARγ的蛋白水平(图5E和5G)。AMPK抑制剂化合物C剂量依赖性地抑制了葡萄糖饥饿诱导的AMPK激活,进而抑制了两种细胞类型中葡萄糖饥饿诱导的TBC1D1磷酸化(图5H和5I)。化合物C剂量依赖性地恢复外源PPARγ2- MYC在葡萄糖饥饿HEK293T细胞中的蛋白水平(图5H)以及内源性PPARγ在葡萄糖饥饿HepG2细胞中的蛋白水平(图5I)。此外,在HepG2细胞中,葡萄糖消耗降低了Rab2A的GTP结合形式,而化合物C逆转了这种效应(图5J)。综上所述,这些数据表明AMPK-TCB1D1-Rab2A轴调节PPARγ蛋白水平以响应营养状况。
6.Rab2A调节细胞脂质积累
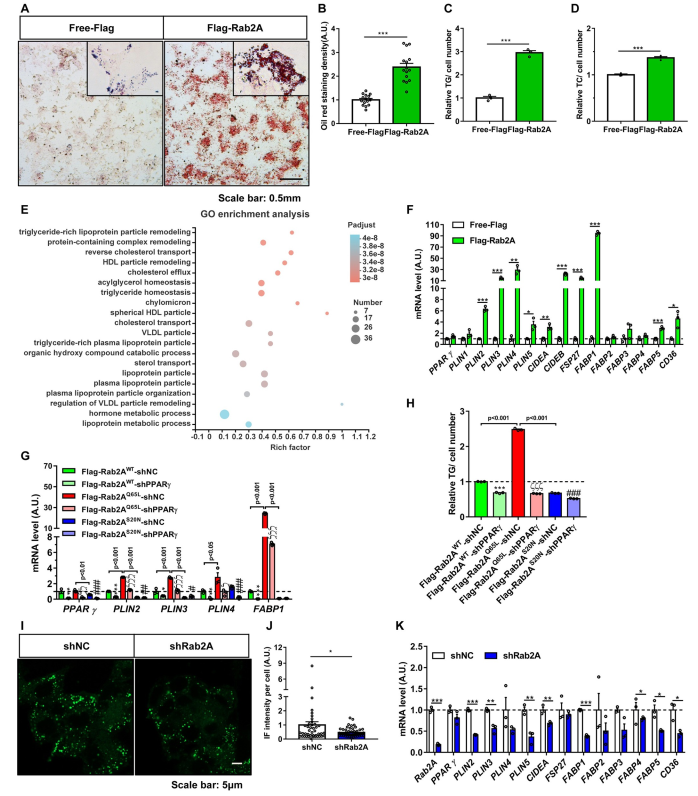
油红O染色显示,Rab2A稳定过表达导致HepG2细胞脂滴积累(图6A和6B)。与对照组相比,过表达Rab2A的HepG2细胞的细胞TG和总胆固醇(TC)水平显著升高(图6C和6D)。基因本体论表达的差异基因主要参与TG稳态和脂蛋白颗粒重塑和运输(图6E)。在稳定过度表达Rab2A的HepG2细胞中,PPARγ靶基因的转录水平显著增加,如PLIN4、CIDEA和FSP27/CIDEC(图6F)。Rab2A对PPARγ靶基因表达及细胞TG积累的影响取决于其鸟嘌呤核苷酸结合状态。与GTP结合的Rab2AQ65L促进PPARγ靶基因的表达(图6G),并引起细胞中TG的积累(图6H)。PPARγ基因的下调抑制了Rab2AQ65L诱导的PPARγ靶基因的表达(图6G),并阻止了Rab2AQ65L诱导的细胞TG积累(图6H)。与过表达Rab2A相比,在HepG2细胞中稳定下调Rab2A可降低细胞脂滴数量(图6I和6J)并抑制PPARγ靶基因的mRNA表达(图6K)。这些数据表明,Rab2A通过PPARγ调控脂质储存基因表达和细胞TG积累。
7.抑制Rab2A可减轻饮食诱导的肝脂积累
通过AAV8介导的shRab2A下调DIO小鼠肝脏中Rab2A的表达。在给予表达shRNA的AAV8 2个月后,对小鼠进行分子和生理分析。小鼠肝脏的分子分析证实了Rab2A在mRNA和蛋白水平上的显著下降(图7A)。不仅shRab2A小鼠肝脏内源性PPARγ2蛋白显著降低,而且这些小鼠的内源性PPARγ1蛋白也观察到较不明显的降低(p = 0.057),同时PPARγmRNA保持正常(图7A和7B)。除了PPARγ蛋白的降低,脂质代谢相关基因和胆固醇合成相关基因在蛋白水平上的表达降低,而FGF21在shRab2A小鼠的肝脏中显著增加(图7A和7B)。shRab2A小鼠肝脏中TG含量显著降低(图7C),shRab2A小鼠肝脏TC水平下降(图7D),shRab2A小鼠血清TG和TC水平均显著降低(图7E和7F)。综上所述,这些数据表明,抑制Rab2A降低了PPARγ蛋白,从而减轻了饮食诱导的体内肝脂积累。

结论:
综上所述,作者发现Rab2A在AMPK-TBC1D1轴下游发挥遗传作用,调控肝脏PPARγ蛋白的稳定性,从而调控PPARγ靶基因的表达,使TG在肝脏中积累,以响应能量/营养状态,这对脂肪肝的治疗有重要意义。