






ACSL4缺乏症对铁死亡介导的急性肾损伤具有保护作用
2012年首次发现铁死亡会导致急性肾损伤(AKI)。但其在AKI中的作用机制尚不清楚。通过对AKI小鼠和正常小鼠肾脏的RNA序列分析,发现酰基-辅酶a合成酶长链家族(ACSL4)差异表达,本研究探讨ACSL4在AKI中的作用。本文于是2022年2月发表于《Redox Biology》,IF=9.986。
本文技术路线:
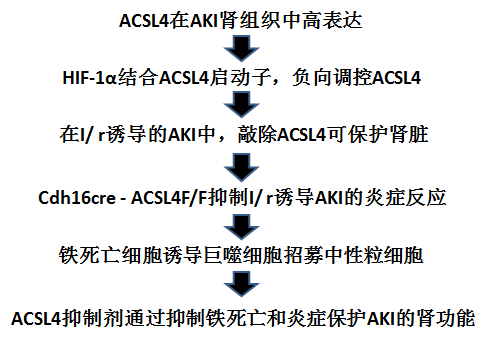
本研究主要内容:
1. ACSL4在AKI肾组织中高表达
RNA-seq测序结果显示,在I/ R诱导小鼠AKI的肾组织中,在铁死亡相关的几条关键通路,如谷胱肽过氧化物酶4 (GPX4)、铁抑制蛋白1 (FSP1)、ACSL4通道等,只有ACSL4表达上调(Fig 1A)。Q-PCR、WB、免疫组化检测ACSL4表达水平。发现ACSL4 mRNA和蛋白质水平在I/R和fa诱导的AKI小鼠肾组织中均上调(Fig. 1B–E)。此外,在FA-AKI和I/R-AKI小鼠中,ACSL4 mRNA水平与BUN和CRE呈显著正相关(Fig. 1F,G)。因此,ACSL4在AKI中呈高表达,并与AKI严重程度呈正相关。

Fig1 AKI患者ACSL4表达上调
2. HIF-1α结合ACSL4启动子,负向调控ACSL4
分析RNA-seq数据分析显示HIF-1信号通路富集(Fig. 2A)。HIF-1信号在AKI中被广泛报道。此外,HIF-1α激动剂可抑制AKI。WB和RT-PCR结果显示,AKI中HIF-1α和HIF-2α表达下调,ACSL4表达上调(Fig. 2B)。然后用低氧(1% O2)诱导HIF表达,探讨其对ACSL4的影响。缺氧可上调HIF-1α和HIF-2α的蛋白和mRNA水平而下调ACSL4的水平(Fig. 2C,D)。此外,shHIF-1α/2α伴侣蛋白ARNT的mRNA水平未被shHIF-1α/2α敲除,而略有升高,可能是代偿性升高。然而,敲低HIF-1α能显著上调ACSL4的表达水平(Fig 2E. F)。同样的,HIF-1α抑制剂BAY 87-2243-a上调ACSL4 mRNA水平。然而,HIF-2α抑制剂PT2385并没有上调ACSL4的mRNA水平(Fig. 2G)。此外,敲低HIF-1α可上调HK-2细胞对铁死亡的敏感性,而抑制ACSL4可逆转其作用(Fig 2H)。HIF-1α基因敲除后,CCL2的mRNA水平升高(Fig. 2I)。用染色质免疫沉淀法探讨HIF-1α的调控机制。首先将HIF-1α过表达质粒转染到HK-2细胞。荧光素酶实验表明HIF-1α抑制ACSL4的表达(Fig. 2J)。然后,在21%或1% O2条件下分离HK-2细胞的细胞质和细胞核。结果发现,HIF-1α和ACSL4分别在细胞核和细胞质中表达(Fig. 2K)。最后,Chip实验发现HIF-1α可以结合ACSL4的启动子并干扰ACSL4的表达(Fig. 2L)。综上所述,HIF-1α可以结合ACSL4启动子,对ACSL4进行负向调控。AKI中下调HIF-1α可导致AKI中ACSL4的高表达,从而诱发铁死亡。
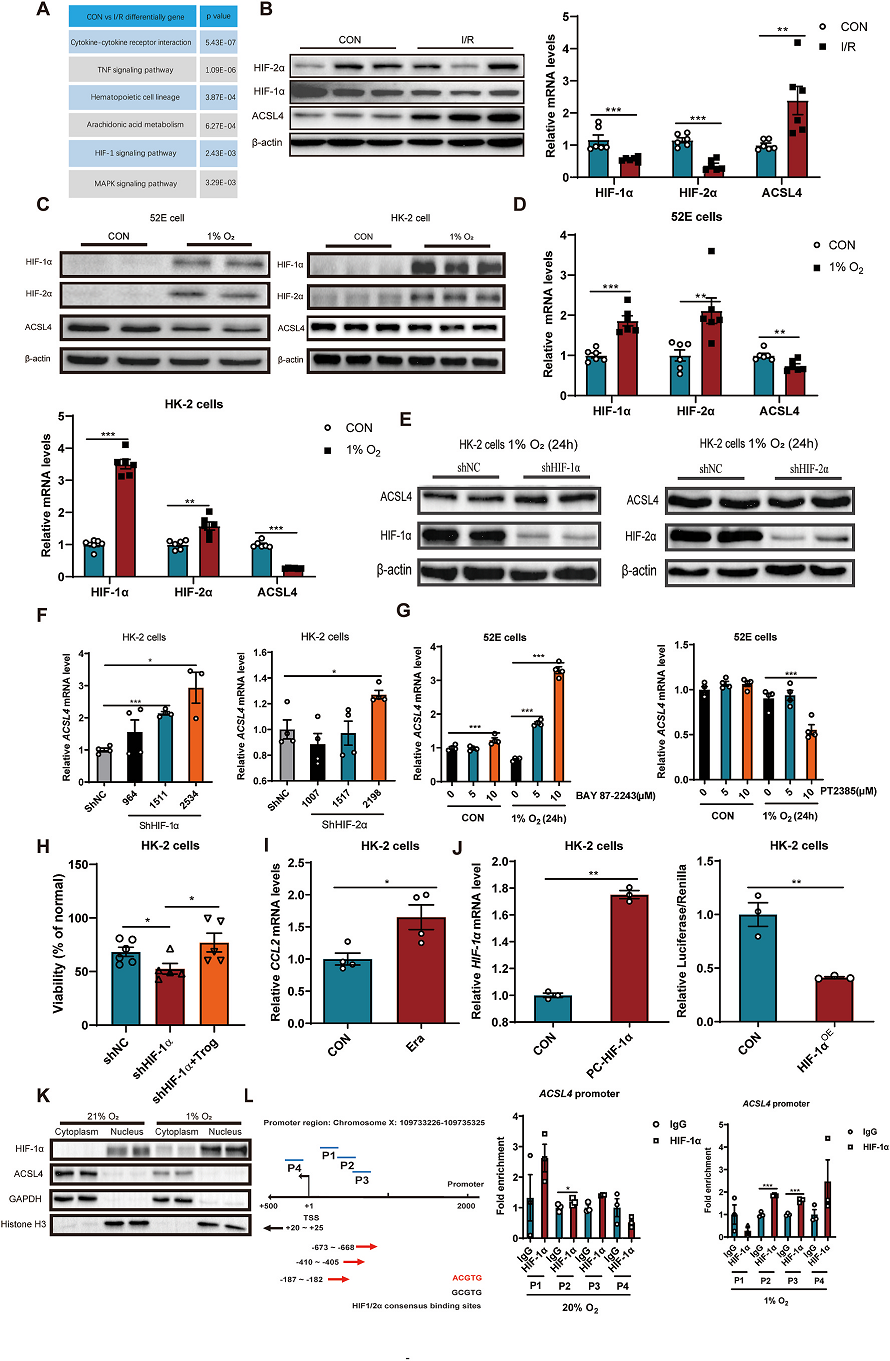
Fig2 HIF-1α结合ACSL4启动子,负向调控ACSL4
3. 在I/ r诱导的AKI中,敲除ACSL4可保护肾脏
建立ACSL4肾小管条件敲除小鼠,研究ACSL4在AKI中的作用。ACSL4基因敲除效果得到证实,结果显示,与ACSL4F/F小鼠相比,Cdh16Cre-ACSL4F/F小鼠肾脏组织中ACSL4蛋白及mRNA表达水平显著下调(Fig. 3A and B)。然后,在ACSL4F/F和Cdh16Cre-ACSL4F / F老鼠用I/ r诱导AKI。免疫荧光染色、免疫组化染色和PCR结果显示,ACSL4在cdh16crea - acsl4f /F小鼠肾脏中的表达水平下调,尤其是在肾小管上皮细胞中,因为ACSL4与肾小管上皮标志物细胞角蛋白18 (CK18)共定位(Fig. 3C-E)。Cdh16Cre-ACSL4F/F小鼠的GPX4和Ptgs2 mRNA表达水平分别上调和下调(Fig. 3E)。GPX4是一种重要的抗氧化过氧化物酶,它的失活直接导致铁死亡。而Ptgs2也是一种已知的铁死亡增加标记物。因此,这些结果证实了Cdh16Cre-ACSL4F/F小鼠对铁死亡的抑制作用。此外,Cdh16Cre-ACSL4F/F小鼠肾脏组织脂质氧化产物MDA含量显著降低(Fig. 3F) 。BUN和CRE含量均显著降(Fig. 3G)。此外,Cdh16Cre-ACSL4F/F小鼠肾脏的病理损伤减轻(Fig. 3H), TUNEL染色的 (Fig.3I)表明ACSL4敲除对I/ r诱导的AKI具有保护作用。因此,在肾小管中敲除ACSL4可以通过靶向铁死亡来抑制I/ r诱导的AKI。

Fig. 3 敲除肾ACSL4对AKI有保护作用
4. Cdh16cre - ACSL4F/F抑制I/ r诱导AKI的炎症反应
炎症也被认为是AKI的关键因素。铁死亡是一种促炎因子,招募巨噬细胞,引起AKI的炎症。RNA序列分析显示,在ACSL4F / F I / R组基因水平显著改变。相比之下,Cdh16Cre-ACSL4F/F组基因水平是正常的(Fig.4A). Cdh16Cre-ACSL4F/F降低了高水平的炎症因子,包括Mmp8, Hmox1, Lcn2,Cxcl2、Cxcl1、Stat3、Cxcl3、IL-6 (Fig. 4B)。RT-PCR结果显示,Cdh16Cre-ACSL4F/F小鼠的肾组织中趋化因子(CCL2)和炎症因子(IL-1β和IL-6) mRNA水平显著降低(Fig. 4B)。RT-PCR结果显示,Cdh16Cre-ACSL4F/F的小鼠肾组织中趋化因子(CCL2)和炎症因子(IL-1β和IL-6) mRNA水平显著降低(Fig. 4C)。流式细胞术分析显示CD11b+F4/80+巨噬细胞在Cdh16Cre-ACSL4F/F小鼠肾脏的浸润明显减少(Fig. 4D)。血常规和免疫组化实验也显示肾脏单核细胞减少,Cdh16Cre-ACSL4F/F小鼠巨噬细胞浸润减少((Fig. 4E,F)。总的来说,敲除ACSL4可以减少I/ r诱导小鼠肾脏中的炎症和巨噬细胞聚集

Fig4敲除肾脏ACSL4可抑制肾脏炎症因子的释放
5. 铁死亡细胞诱导巨噬细胞招募中性粒细胞
AKI小鼠中性粒细胞趋化因子显著升高,Cdh16Cre-ACSL4F/F小鼠中性粒细胞趋化因子显著降低(Fig.5A)。流式细胞术、血常规及免疫组化实验证实了中性粒细胞中Cdh16Cre-ACSL4F/F下调作用(Fig. 5B–D)。因此,作者利用铁细胞和中性粒细胞进行体外趋化实验,以确定中性粒细胞是否也被铁死亡细胞招募。然而,嗜中性粒细胞不能被铁死亡细胞吸收(Fig. 5E). 此外,铁死亡细胞不诱导趋化因子的产生(Fig. 5F)。研究表明,巨噬细胞释放趋化因子招募粒细胞到损伤部位。由于铁细胞诱导的巨噬细胞CXCL1和CXCL2的表达水平上调,巨噬细胞可能会诱导巨噬细胞招募中性粒细胞(Fig. 4H)。体外趋化实验也显示嗜中性粒细胞被巨噬细胞招募(Fig. 5G)。此外,嗜铁抑制剂Fer-1下调CXCL1的表达水平, CXCL2及肾脏中性粒细胞浸润(Fig. 5H,I)。因此,铁细胞间接招募中性粒细胞,提供了铁细胞在AKI中的另一种促炎作用。尽管如此,cdh16crea - acsl4F/F能有效抑制这种促炎作用。

Fig.5巨噬细胞通过分泌中性粒细胞趋化因子来招募中性粒细胞
6. ACSL4抑制剂罗格列酮(Rosi) 通过抑制铁死亡和炎症保护AKI的肾功能
ACSL4抑制剂Rosi用于进一步验证ACSL4在AKI中的作用。结果发现,I/R+Rosi组降低了血液CRE、肾脏指数,减轻了病理性肾脏损伤(Fig. 6A–C)。此外,Rosi下调了ACSL4、CCL2、CXCL1和CXCL2的水平(Fig. 6D)。免疫组织化学和流式细胞术显示I/R+Rosi组肾巨噬细胞浸润较少(Fig. 6E,F)。总的来说,这些发现表明Rosi对AKI有保护作用,减少炎症细胞的浸润,提示ACSL4在AKI中发挥重要作用。
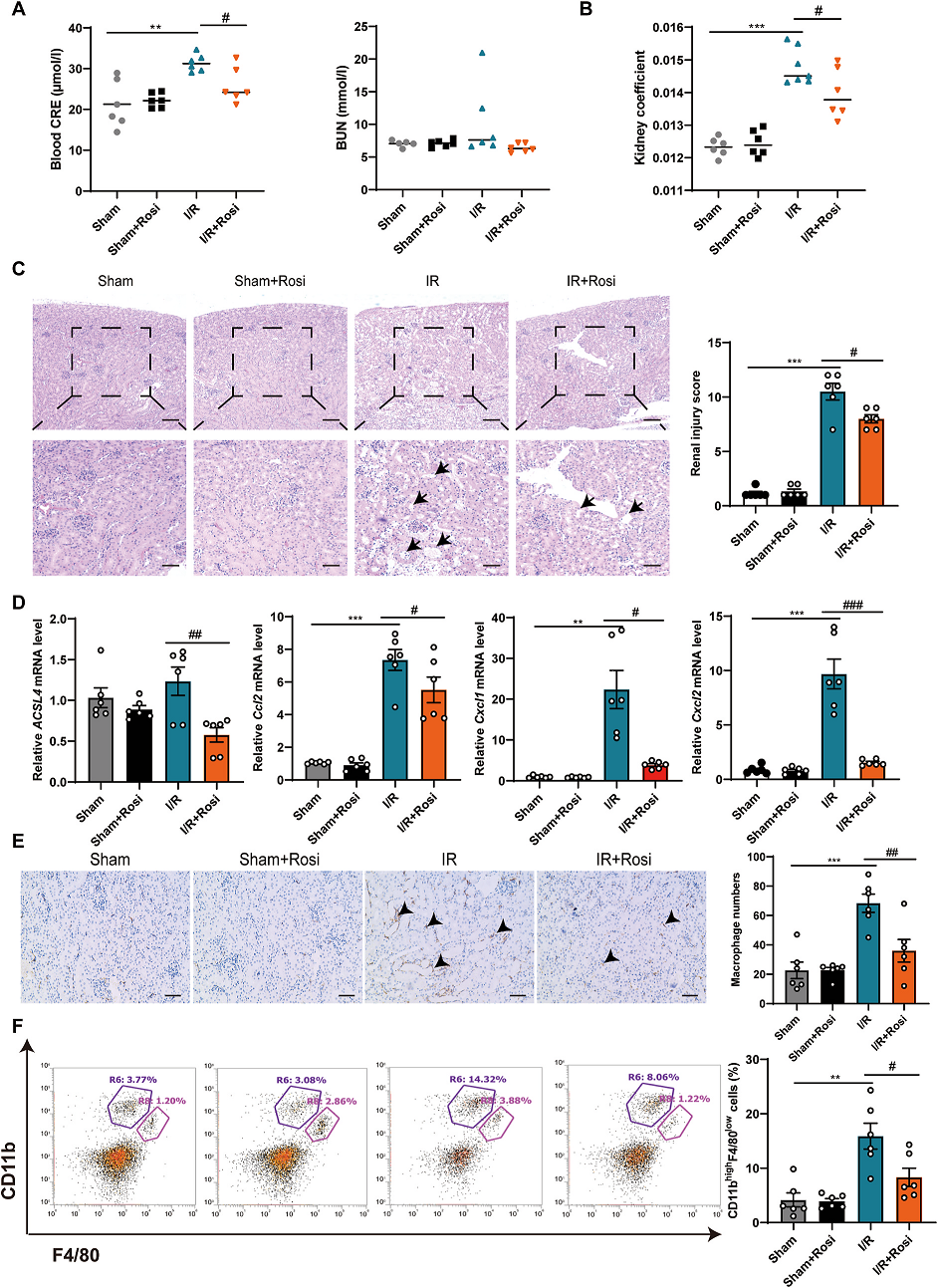
Fig. 6. ACSL4抑制剂罗格列酮(Rosi)通过抑制铁下垂和炎症来保护AKI患者的肾功能
本研究发现,ACSL4是通过抑制铁下垂来预防和治疗AKI的潜在靶点。HIF-1α通过结合ACSL4启动子,负向调控ACSL4的表达;铁细胞招募巨噬细胞并刺激巨噬细胞招募中性粒细胞,从而引起AKI的炎症级联反应。本研究结果为AKI和铁死亡相关疾病新治疗方法的开发提供了参考。
参考文献:
[1] Wang Y, Zhang M, Bi R, et al. ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury[J]. Redox Biology, 2022: 102262.