






HMGA2通过肿瘤相关巨噬细胞招募促进结直肠癌进展
肿瘤相关巨噬细胞(TAMs)通常表现为肿瘤前M2样表型,通过其免疫抑制活性强烈影响结直肠癌(CRC)的进展。HMGA2是一种癌蛋白,在CRC细胞中异常过表达。然而,肿瘤来源的HMGA2调节结直肠癌肿瘤微环境的机制尚不清楚。我们发现,在体外和体内,癌细胞中的HMGA2促进巨噬细胞招募和M2极化。HMGA2直接与STAT3启动子结合激活其转录,进而诱导CCL2分泌,从而促进巨噬细胞募集。肿瘤细胞中HMGA2的表达和间质中CD68的表达之间存在很强的正相关。在所有患者或远处转移阴性的亚组中,CD68表达升高的患者的总生存期较差。我们的工作揭示了CRC中HMGA2/STAT3/CCL2轴和巨噬细胞招募之间的联系。这些发现为靶向CRC中的HMGA2/STAT3/CCL2轴提供了一种新的治疗选择。本文于2022年1月发表于“Theranostics”(IF=11.556)。
技术路线

结果
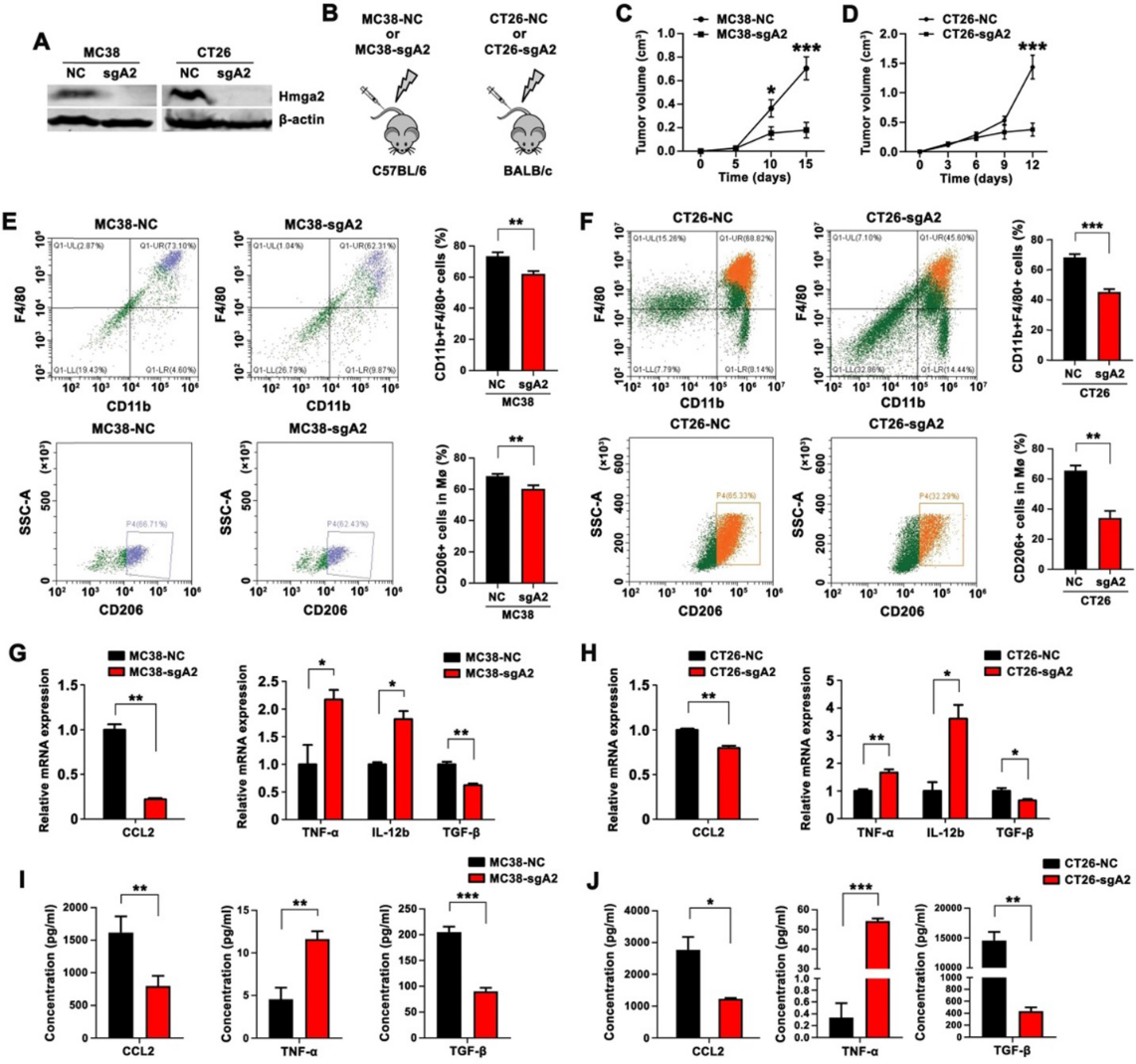
1)在CRC细胞中敲除HMGA2可抑制TAM浸润、M2极化和CCL2的分泌
利用CRISPR/Cas9技术,我们用特异性sgRNA在小鼠CRC细胞系(MC38和CT26)中产生了稳定的Hmga2敲除(Hmga2- KO)细胞。Western blot分析验证了敲除Hmga2的有效性(图1A)。C57BL/6小鼠皮下接种对照或Hmga2-KO MC38细胞(MC38-NC和MC38-sgA2),BALB/c小鼠皮下接种CT26-NC或CT26-sgA2细胞(图1B)。结果显示,sgRNA介导的敲除Hmga2在C57BL/6和BALB/c皮下肿瘤模型中均显著抑制肿瘤生长(图1C-D)。
M2巨噬细胞可以促进肿瘤的生长和发展。为了阐明Hmga2在TAM招募和极化中的作用,我们采用流式细胞术量化巨噬细胞(CD11b+F4/80+)和M2巨噬细胞(CD11b+F4/80+CD206+)的百分比。携带Hmga2敲除MC38肿瘤的小鼠,浸润性CD11b+F4/80+巨噬细胞和CD11b+F4/80+CD206+ M2巨噬细胞减少(图1E),提示Hmga2敲除的抗肿瘤作用可能与CRC中TAMs的招募和极化有关。同样,在Hmga2敲除CT26肿瘤的小鼠中也观察到了类似的结果(图1F)。这些结果表明,Hmga2在体内促进TAM招募和M2极化。
CCL2是一种重要的趋化因子,有助于巨噬细胞招募和浸润。为了探究CCL2是否受Hmga2调控,我们采用qPCR检测CCL2的表达,ELISA检测CCL2的分泌。如图1G和1I所示,与对照组相比,MC38-sgA2细胞肿瘤中CCL2的表达减少,产生的CCL2减少。同样,通过qPCR(图1H)和ELISA检测(图1J),在CT26皮下肿瘤模型中,Hmga2的缺失抑制了CCL2的表达和分泌。
接下来,为了探讨TME中CRC细胞中Hmga2缺失与巨噬细胞极化之间的关系,我们评估了M1相关(TNF-α和IL-12b)和M2相关(TGF-β)细胞因子的表达。如图1G-H所示,qPCR结果显示,CRC细胞敲除Hmga2后,MC38和CT26异种移植瘤中TNF-α和IL-12b的表达均增加,TGF-β水平明显降低。在MC38和CT26皮下肿瘤模型中,ELISA进一步证实Hmga2-KO肿瘤组织中TNF-α增加和TGF-β分泌减少(图1I-J)。总的来说,在CRC细胞中敲除Hmga2可以抑制TAM浸润、M2极化和CCL2的分泌。
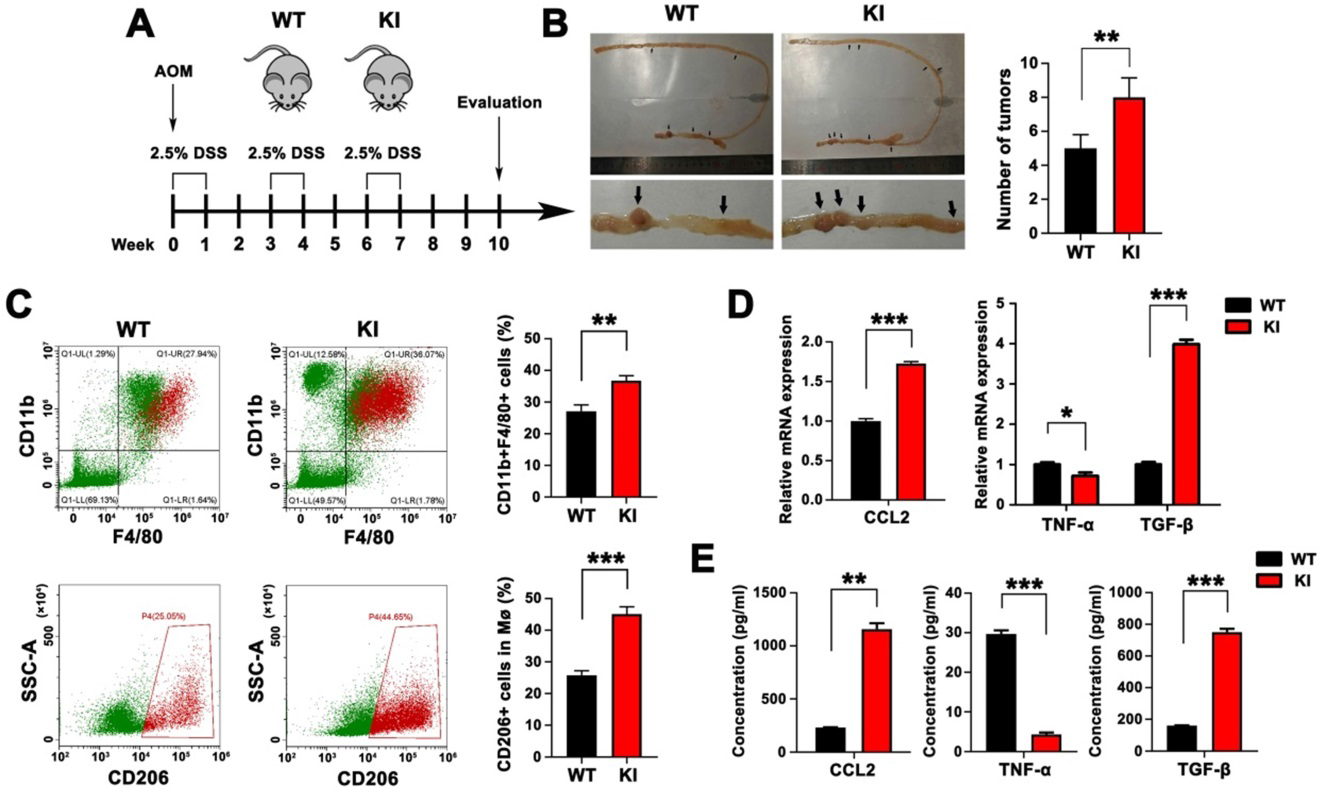
2)在AOM/DSS模型中,HMGA2肠上皮特异性敲入促进TAM浸润、M2极化和CCL2分泌
我们试图了解在CRC肿瘤发生过程中,Hmga2的肠上皮特异性敲入(KI)在TAM浸润和M2极化中的参与。我们使用WT和肠上皮特异性Hmga2 KI小鼠,随后用AOM和DSS治疗诱导结直肠肿瘤(图2A)。如图2B所示,Hmga2 KI小鼠比WT小鼠在肠道内发生的肿瘤更多。与WT小鼠相比,在给药AOM/DSS后,Hmga2 KI小鼠肠道组织中CD11b+F4/80+巨噬细胞和CD11b+F4/80+CD206+ M2巨噬细胞的比例增加(图2C)。此外,通过qPCR和ELISA检测介导巨噬细胞浸润的趋化因子CCL2水平(图2D-E)。我们发现Hmga2 KI可显著上调小鼠肠道组织中CCL2的产生。qPCR结果显示,AOM/DSS处理后,Hmga2 KI小鼠肠道组织中M1细胞因子(TNF-α)的表达显著降低,M2细胞因子(TGF-β)的表达显著增加(图2D)。这些结果经ELISA证实(图2E)。综上所述,肠道上皮特异性Hmga2 KI通过促进TAM浸润、M2极化和CCL2分泌来调节TME。
3)HMGA2促进体外巨噬细胞招募、M2极化和CCL2分泌
为了进一步说明HMGA2在体外是否有助于TAM招募,我们利用Transwell共培养系统。如图3A所示,PMA分化的THP1人单核细胞与HMGA2过表达或不表达的HT29人结直肠癌细胞(HT29-NC和HT29-A2)共培养,RAW264.7细胞与HMGA2敲除或不敲除的CT26小鼠结直肠癌细胞(CT26-NC和CT26-sgA2)。有趣的是,THP1与HT29-A2共培养比HT29-NC细胞表现出更高的迁移能力(图3B)。相反,与CT26-sgA2细胞共培养时,RAW264.7的迁移能力显著减弱(图3C)。随后,我们通过qPCR检测CRC细胞中CCL2的表达水平,发现HMGA2过表达会上调HT29细胞中CCL2的表达(图3D),而HMGA2 KO会下调CT26细胞中CCL2的表达(图3E)。此外,HT29-A2条件培养基培养的THP1细胞中TNF-α的表达下降,而TGF-β则呈现相反的趋势(图3D)。与对照组相比,CT26-sgA2条件培养基处理RAW264.7细胞后,TNF-α水平升高,TGF-β水平降低(图3E)。这些研究进一步证实HMGA2在体外促进巨噬细胞招募、M2极化和CCL2分泌。

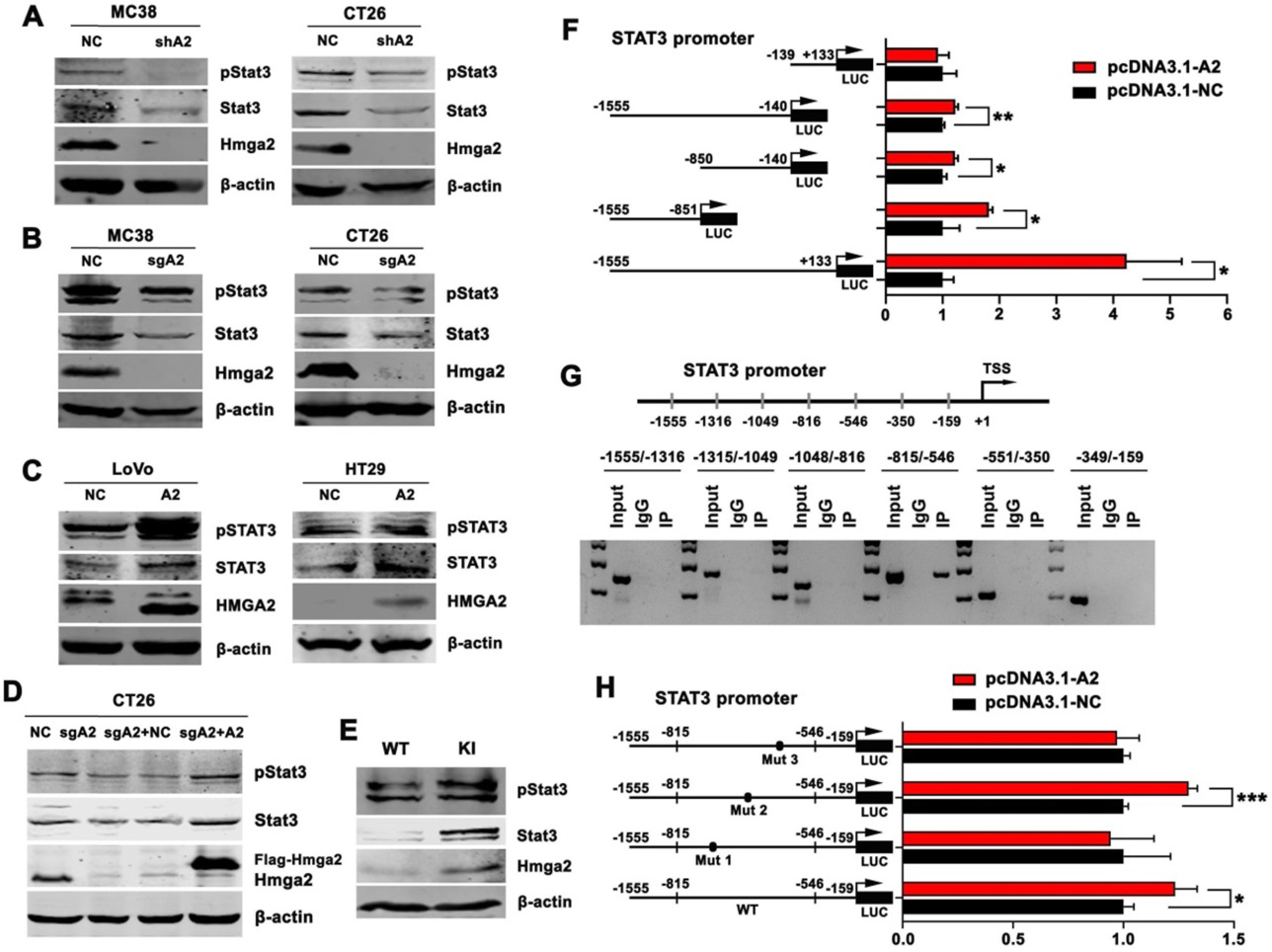
既往研究表明,STAT3在肿瘤免疫耐受中起关键作用。为了研究HMGA2是否通过STAT3依赖机制调控TME中TAMs的免疫抑制,我们分析了CRC中HMGA2与STAT3的关系。如图4A所示,shRNA介导的Hmga2敲低导致MC38和CT26细胞中总和磷酸化的Stat3 (pStat3Tyr705)蛋白水平下降。Western blotting结果显示,sgRNA介导的敲除Hmga2显著抑制了MC38和CT26细胞中Stat3和pStat3Tyr705的表达(图4B)。相反,与对照组相比,Hmga2过表达的LoVo和HT29细胞中STAT3和pSTAT3Tyr705的表达上调(图4C)。此外,我们将对照或过表达Hmga2的载体转染到对照或Hmga2缺失的CT26细胞中(CT26-NC、CT26-sgA2、CT26- sgA2 +NC和CT26-sgA2 +A2)。如图4D所示,Hmga2过表达恢复后,敲除Hmga2后减少的Stat3和pStat3Tyr705的表达量逆转。为了更好地了解Hmga2和Stat3在体内的调控机制,我们通过Western blotting检测了在WT和KI小鼠肠道组织中Stat3、pStat3Tyr705和Hmga2的表达。如图4E所示,我们发现Hmga2的敲入提高了Stat3和pStat3Tyr705的水平。
为了确定HMGA2是否能在转录水平激活STAT3,我们进行了荧光素酶和ChIP检测。如图4F所示,我们将5个人类STAT3启动子片段克隆到pGL3载体上,分别为-139/+133、-1555/-140、-850/-140、-1555/-851和-1555/+133。结果显示,HMGA2过表达显著刺激了STAT3启动子区域-1555/-140、-850/-140、-1555/-851和-1555/+133的荧光素酶活性,说明-1555/-140片段可能参与了HMGA2调控STAT3转录的过程(图4F)。为了进一步证实,我们采用ChIP实验验证了其直接调控机制,并确定了结合位点的位置。如图4G所示,HMGA2直接与STAT3启动子结合,且HMGA2结合位点主要位于-815至-546之间的STAT3启动子区域。接下来,我们分别突变了STAT3启动子段侧翼的三个Hmga2结合位点(-815/-546)。图4H显示的结果表明,转染含有突变1(-743/-730,从TAATTACTCTATTT到TAGCCACTCTACGT)和突变3(-585/-576,从TATCTAACTA到TCTCGCTA)的结构体后,荧光素酶的诱导活性显著减弱。这些结果表明,HMGA2通过直接结合STAT3的-743/-730和-585/-576启动子区域,增强了STAT3的转录。
5)CRC细胞中HMGA2过表达增强CCL2表达和巨噬细胞迁移增加依赖于STAT3
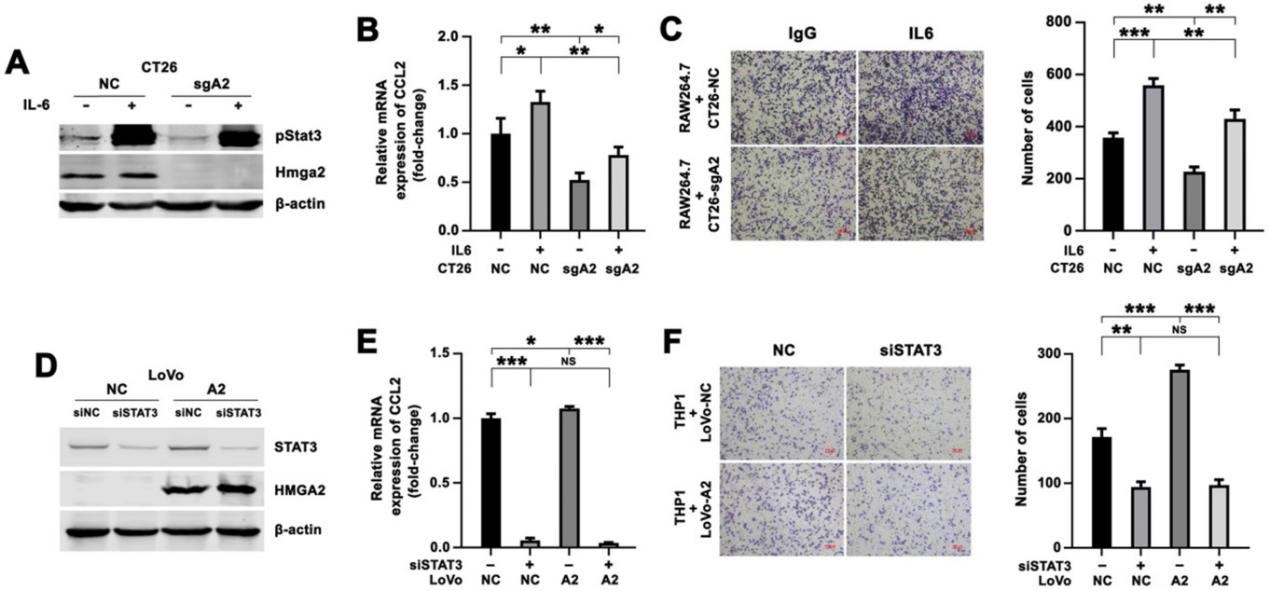
我们通过IL6处理CT26-NC和CT26-sgA2细胞,刺激Stat3,然后与RAW264.7细胞共培养,确定Hmga2是否以Stat3依赖的方式调控CCL2的表达。IL6处理导致pStat3Tyr705表达增加(图5A), CRC细胞中CCL2表达强烈上调(图5B), RAW264.7细胞迁移增强(图5C)。此外,将靶向STAT3的siRNA引入LoVo-NC和LoVo-A2细胞,通过Western blotting评价STAT3抑制效果(图5D)。我们观察到,在LoVo-NC细胞中,siSTAT3组CCL2的表达低于对照组,说明STAT3上调了CCL2的表达(图5E)。我们还发现HMGA2过表达导致CCL2的表达增加,但引入靶向STAT3的siRNA后,CCL2的诱导被取消(图5E)。在Transwell共培养体系中观察到一致的结果。LoVo细胞中STAT3沉默导致THP1细胞迁移减少(图5F)。与LoVo-NC细胞共培养相比,THP1细胞与LoVo-A2细胞共培养显示出更强的迁移能力,但通过引入STAT3 siRNA,这种增加的迁移被消除(图5F)。这些结果表明,HMGA2上调了CRC细胞中CCL2的表达,并以Stat3依赖的方式促进巨噬细胞的迁移。
6)CRC细胞中HMGA2过表达导致巨噬细胞迁移的增加依赖于CCL2
为了研究CCL2在巨噬细胞招募中的作用,我们用重组小鼠CCL2处理CT26-NC和CT26-sgA2细胞,然后与RAW264.7细胞共培养。敲除Hmga2抑制了RAW264.7细胞的迁移潜能,而重组CCL2蛋白处理增强了其迁移潜能(图6A)。此外,我们用中和性抗CCL2抗体预孵育LoVo细胞,然后使用Transwell共培养体系评估THP1细胞的迁移能力。如图6B所示,我们的结果表明,HMGA2过表达促进THP1细胞的迁移,而抗CCL2抗体的处理则使THP1细胞迁移消失,说明HMGA2和CCL2在巨噬细胞招募中起着至关重要的作用。我们还发现,中和CCL2后,与LoVo-A2共培养的THP1迁移诱导被取消。这些数据表明,HMGA2过表达以CCL2依赖的方式促进巨噬细胞的迁移。

7)人结直肠癌标本中HMGA2和CD68表达的临床意义
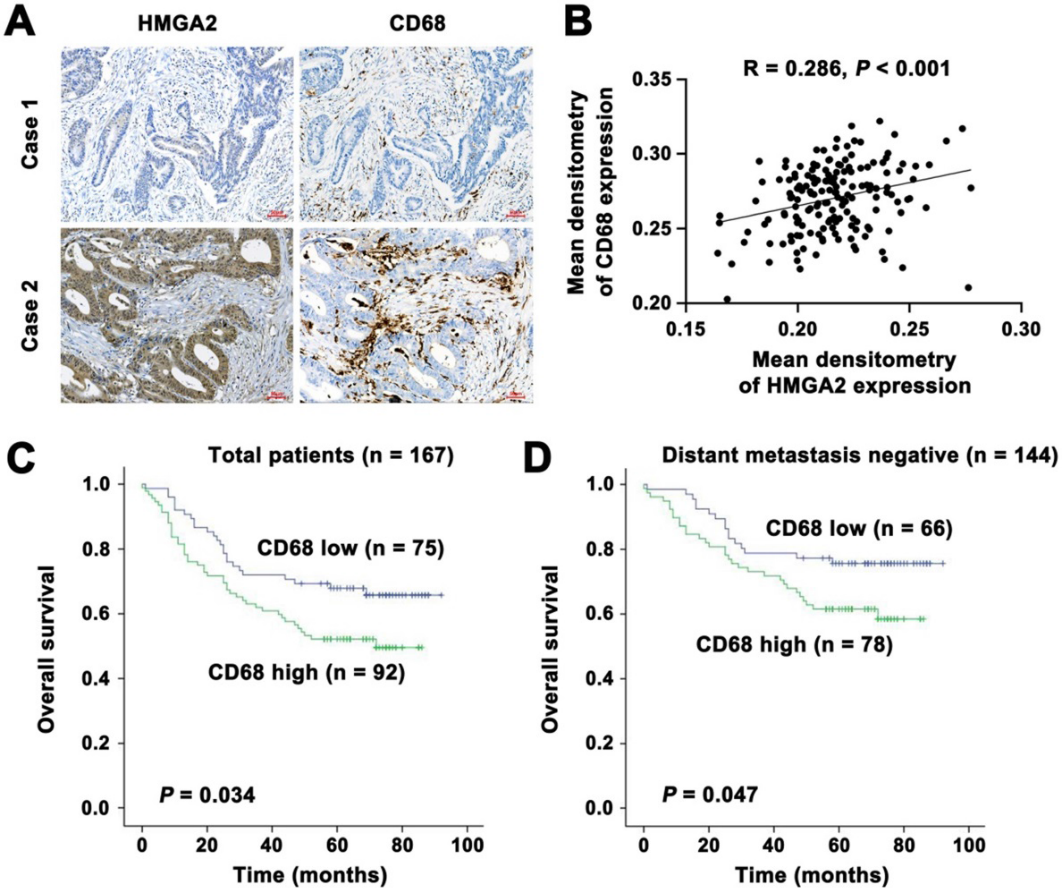
有报道称CD68是巨噬细胞的免疫组化染色标记物。为了研究HMGA2水平与CRC患者巨噬细胞浸润的关系,我们采用免疫组化染色对167例人类CRC标本中HMGA2和CD68的表达进行了评估。结果表明,肿瘤细胞中HMGA2的表达与间质中CD68的表达有很强的相关性。如图7A-B所示,我们发现HMGA2与CD68表达呈正相关趋势。接下来,我们进行Kaplan-Meier生存分析来评估CD68作为CRC预后标志物的潜在价值。如图7C所示,在所有患者中,间质中CD68高表达与患者生存期降低相关。当分层到远处转移阳性和阴性亚组时,我们发现CD68高表达患者的总生存期较差,而CD68低表达患者的总生存期较好(图7D)。这些结果提示间质CD68可作为临床预后的标志和预测因子,提示其在结直肠癌中的临床意义。
结论:CRC细胞中过表达HMGA2通过上调Sat3介导的CCL2分泌,促进巨噬细胞招募和M2极化,从而促进CRC肿瘤免疫抑制。我们的研究揭示了HMGA2在免疫抑制微环境形成中的一种新的促癌作用。
参考文献:
Xin Wang, Jian Wang, Jiahui Zhao, Hao Wang, Jing Chen, Jingjing Wu. HMGA2 facilitates colorectal cancer progression via STAT3-mediated tumor-associated macrophage recruitment. Theranostics 2022; 12(2): 963-975. doi:10.7150/thno.65411.