






iASPP抑制gp78介导的TMCO1降解,维持Ca2+稳态,控制肿瘤生长和耐药
从细胞增殖到凋亡,Ca2+从内质网释放是调节Ca2+稳态的一个重要事件,由多种生物过程协调。Ca2+稳态失调与各种癌症特征有关。本研究探讨已报道的Ca2+通道蛋白TMCO1在结肠癌组织中的作用机制。本研究于2022年1月发表《Proceedings of the National Academy of Sciences of the United States of America》,IF=9.412。
本文技术路线:

1、TMCO1蛋白在结肠癌中高表达
在肺腺癌(LUAD)、子宫内膜癌(UCEC)、胃腺癌(STAD)和结肠腺癌(COAD) 等不同类型的癌症中(Figure 1A),TMCO1的含量显著增加。其中COAD表现出最高的表达量变化(Figure 1A)。Western blot (WB)进一步验证了40对结肠癌组织及癌旁组织,发现TMCO1蛋白在结肠癌组织中上调(Figure 1B)。作者还发现,TMCO1蛋白的高表达与结肠癌晚期显著相关,但与性别和年龄无关(Figure 1C,D)。作者的RT-PCR结果显示,TMCO1 mRNA在结肠癌中也没有明显变化(Figure 1E)。为了探讨这一点,作者采用COAD作进一步分析。与体内结果一致的是,TMCO1蛋白在蛋白水平上增加,而在mRNA水平上没有增加(Figure. 1F)。作者还发现蛋白酶体抑制剂MG132,而不是溶酶体抑制剂氯喹(CQ),提高了TMCO1蛋白的表达,提示TMCO1可以被蛋白酶体降解所调节(Figure. 1G),H)。已有研究发现,目标蛋白的蛋白酶体降解主要由多聚泛素化介导。与此一致,TMCO1被发现具备多泛素化特征(Figure. 1I)。
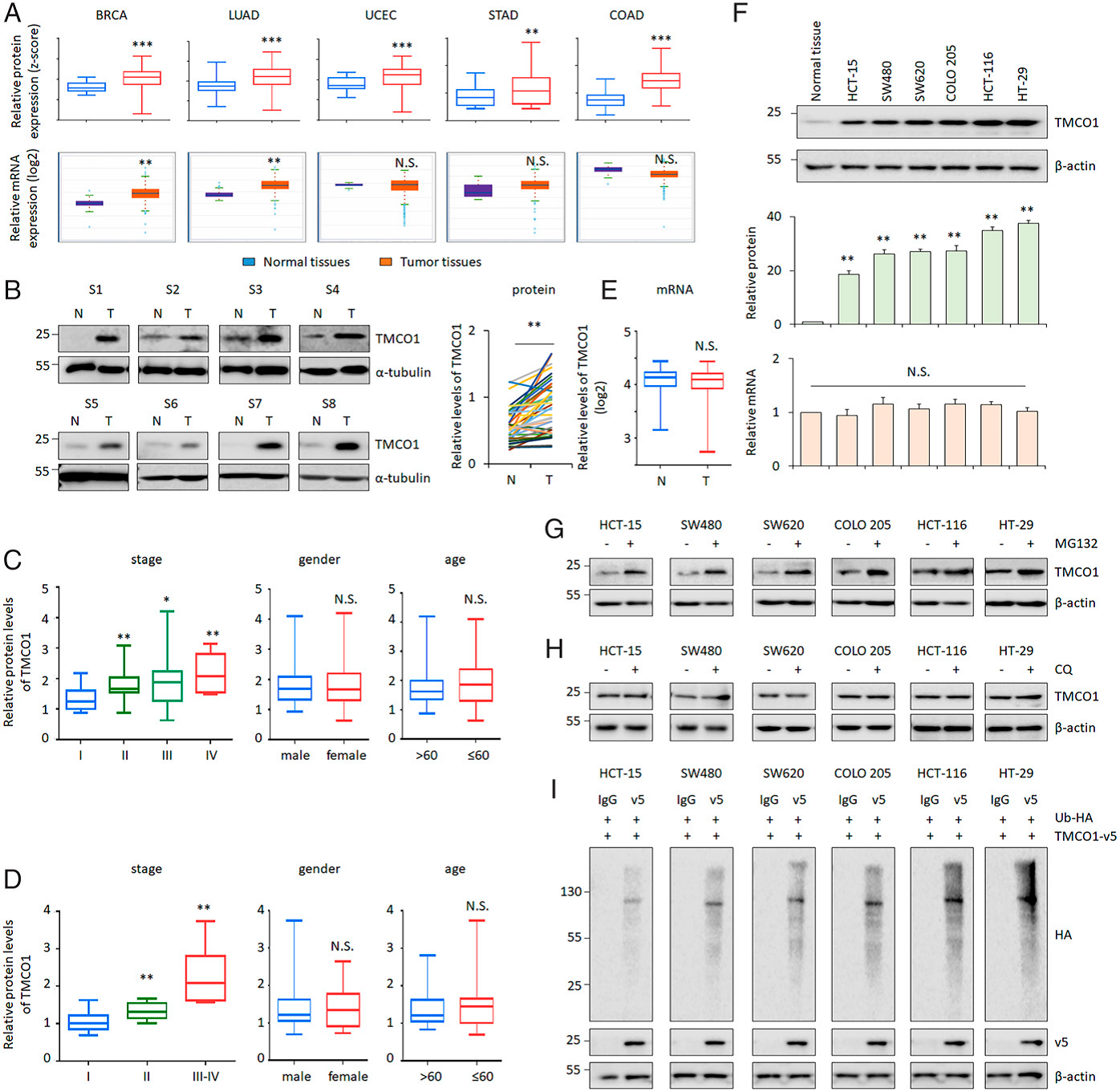
Fig 1 TMCO1蛋白在人结肠癌组织中过表达
2.TMCO1是E3连接酶Gp78的底物
作者想知道TMCO1蛋白的变化是否影响其在COAD中的激活。为了验证这一点,作者首先探索了可能参与调控TMCO1蛋白稳定性的E3连接酶,通过抑制ER定位的E3连接酶的表达来测定其蛋白水平。结果表明,结果表明,Gp78、RNF5、RNF185基因敲除可增加TMCO1的表达,而HRD1、SPFH1、TMEM129基因敲除则不能增加TMCO1的表达(Figure 2A)。IP检测显示,Gp78存在于外源表达的TMCO1沉淀中,而RNF185和RNF5都不可能与TMCO1结合(Figure 2B)。TMCO1与Gp78的直接相互作用进一步在体外得到验证 (Figure 2C)。因此,作者推断TMCO1可能是Gp78的底物。
Gp78 过表达降低了TMCO1蛋白的稳态水平,而加入MG132后,TMCO1蛋白的稳态水平没有进一步降低证实了这一观点(Figure 2D)。此外,野生型(WT) Gp78泛素化TMCO1(Figure 2E)。与此一致,缺陷的K48R泛素突变体阻止了TMCO1泛素链的形成(Figure 2F)。此外, siRNA介导了Gp78KD几乎完全消除了TMCO1 K48的泛素化
通过生物信息学预测工具,发现K40、K116、K160、K172和K186最有可能是被泛素化修饰的位点。为了验证生物信息学结果,作者构建了泛素化缺陷TMCO1突变体(K40R、K116R、K160R、K172R和K186R)。分别将单个突变体引入HT-29细胞后进行泛素化实验。如图所示,K186R-TMCO1基本丧失了被泛素化的能力,其他突变体的泛素化程度与WT TMCO1相似(Figure 2G)。环己酰亚胺(CHX)被广泛用于抑制真核细胞的蛋白质合成。经CHX处理后,TMCO1在对照细胞中的半衰期为4 h,K186R-TMCO1比野生型TMCO1更长(Figure 2H)。而Gp78未降低K186R-TMCO1的表达(Figure 2I)。说明Gp78在K186位点催化TMCO1的泛素化,导致其泛素介导的蛋白酶体降解。
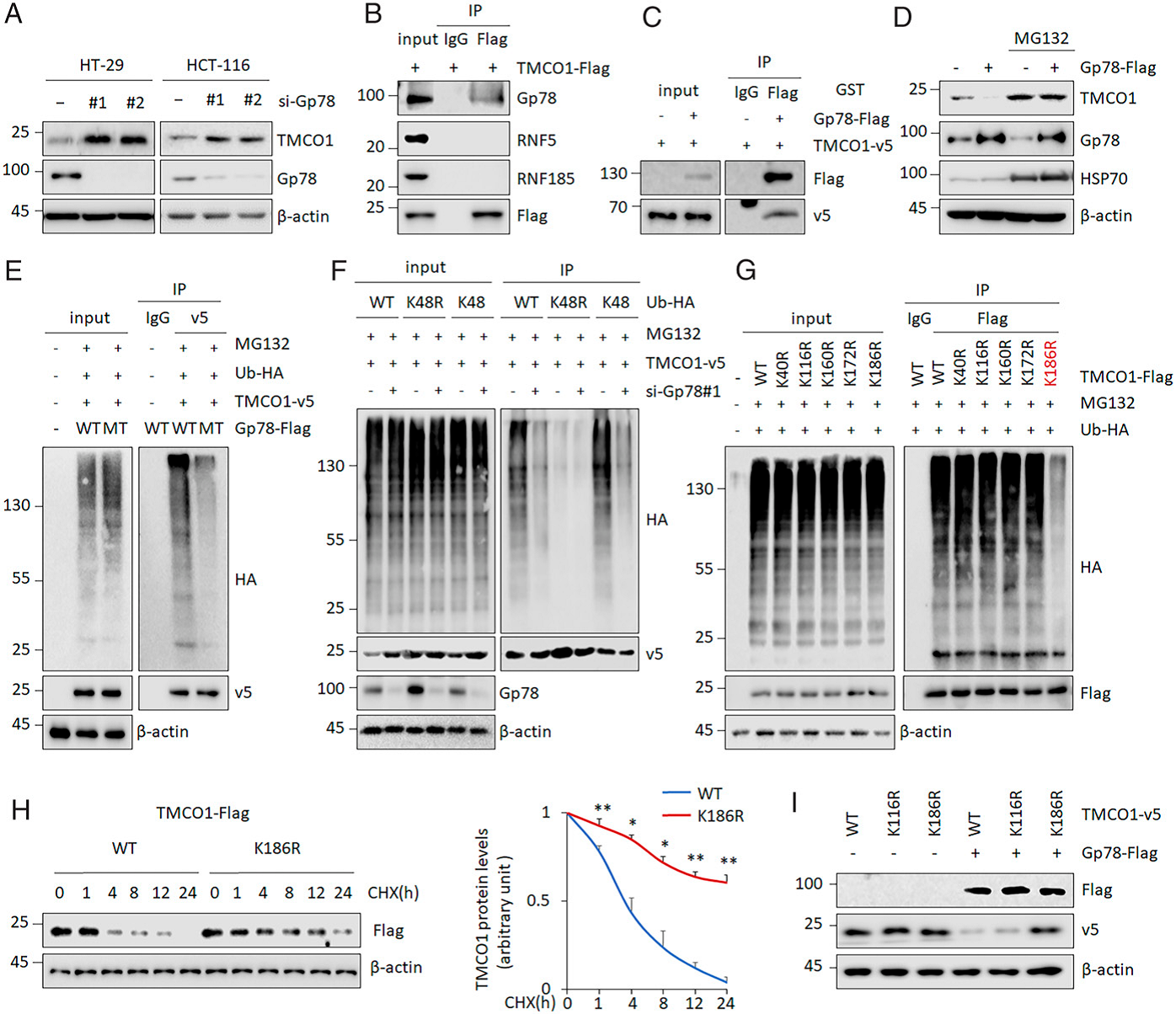
Fig 2 TMCO1是Gp78的底物
3、在结肠癌组织中,癌基因iASPP和TMCO1相互关联
通过WB,检测发现Gp78在结肠癌标本与配对的相邻正常对照的表达水平无明显差异(Figure 3A,B)。同时检测Gp78和TMCO1,发现两者之间没有明显的相关性(Figure 3C)。作者使用anti-Gp78下拉进行质谱分析,寻找Gp78相互作用的蛋白。结果发现一个众所周知的致癌基因iASPP(Figure 3D)。作者还发现,与相邻的正常对照组相比,iASPP在结肠癌组织中增加(Fig. 3E)。与TMCO1一样,iASPP表达与晚期结肠癌相关(Figure 3F)。此外,TMCO1的增加与iASPP的增加呈正相关(Figure 3G),说明iASPP的表达与TMCO1在体内的表达有关。
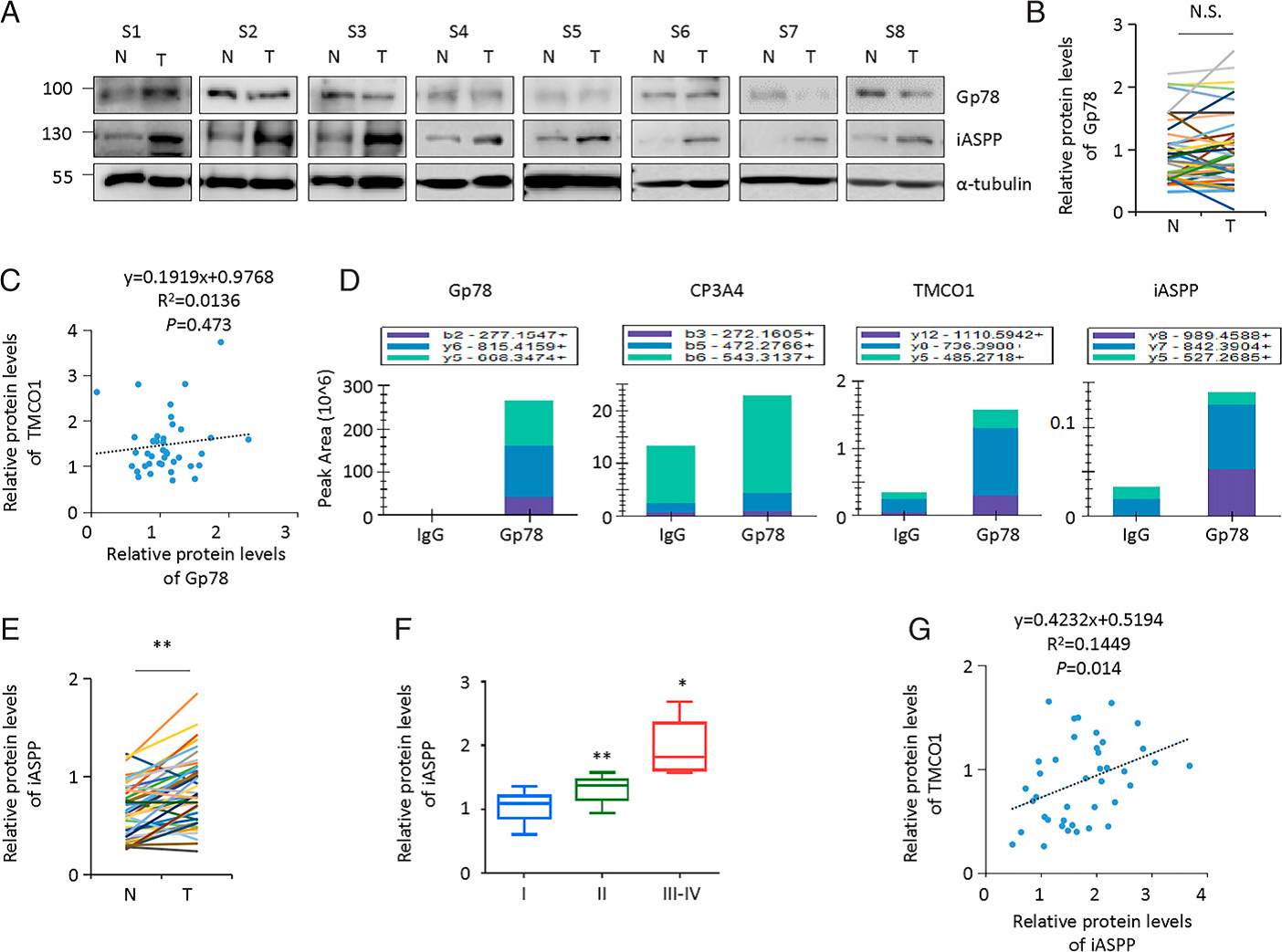
Fig 3癌基因iASPP和TMCO1在结肠癌组织体内相互关联
4、iASPP保护TMCO1免受泛素-蛋白酶体降解
用iASPP 过表达或敲低检测结肠癌细胞系中TMCO1的水平。值得注意的是,当iASPP过表达时,TMCO1持续且显著增加,iASPP敲低将会抑制TMCO1表达(Figure 4A)。但 iASPP对TMCO1 mRNA表达没有影响(Figure 4A)。而MG132却不能完全拯救iASPP 敲低引起的TMCO1抑制(Figure 4B)。与对照组4个小时的半衰期相比,iASPP过表达的细胞半衰期延长到12小时左右,而iASPP敲低的细胞在半衰期缩短至1小时左右(Figure 4C,D)。此外,iASPP 过表达抑制TMCO1多聚泛素化,而iASPP 敲低则增加了TMCO1多聚泛素化(Figure 4E,F)。这些数据共同表明,致癌基因iASPP保护TMCO1免受泛素介导的蛋白酶体降解。此外,iASPP调控的TMCO1表达被Gp78 敲低完全消除(Fig. 4G)。这并不是由于iASPP蛋白本身的变化,因为Gp78kd对iASPP表达无明显影响(Fig. 4G)。此外,与WT TMCO1及泛素化位点无关TMCO1突变体(K116R)相比,泛素化缺陷TMCO1 K186R对iASPP介导的TMCO1增加具有高度耐药性(Fig. 4H)。因此,iASPP通过依赖Gp78的方式稳定TMCO1。

Fig 4 iASPP保护TMCO1免受泛素介导的蛋白酶体降解
5、iASPP通过与Gp78竞争性结合稳定TMCO1
作者观察到Gp78、TMCO1和iASPP形成复合物(Figure 5A),这与之前的质谱结果一致。此外,iASPP 过表达降低了Gp78与TMCO1的结合(Figure 5A)。因此,有理由认为iASPP可能通过抑制Gp78与TMCO1的结合来发挥作用。如果是这种情况,iASPP可能与Gp78或TMCO1相关联。为验证这一观点,使用体外翻译或重组的iASPP、Gp78和TMCO1蛋白进行体外结合实验。结果表明,iASPP与Gp78共沉淀,只有Gp78存在时,iASPP才与TMCO1发生相互作用(Fig. 5B)。因此,说明iASPP直接与Gp78相互作用,而不与TMCO1相互作用。
作者构建了4个Gp78截断突变体,如图5C所示。一个抗v5标签抗体的体外结合实验显示v5标记的TMCO1与WT-和F2(301-400)-共免疫沉淀,且两者都含有Ring结构域,而没有其他结构域,表明Ring结构域对于Gp78与TMCO1的结合是必不可少的(Fig. 5D)。通过体外IP分析,作者发现(301-400)Gp78,即与TMCO1结合的区域,也有助于与iASPP结合(Figure 5E),进一步支持了iASPP与TMCO1在Gp78结合方面存在竞争的假设。在iASPP蛋白增加的情况下比较TMCO1和Gp78两者之间的结合能力。发现两随着iASPP与Gp78结合程度的增加,TMCO1和Gp78逐渐降低(Fig. 5F)。这些数据表明,iASPP与TMCO1竞争获得与(301-400)Gp78结合,增加TMCO1稳定性(Fig. 5G)。
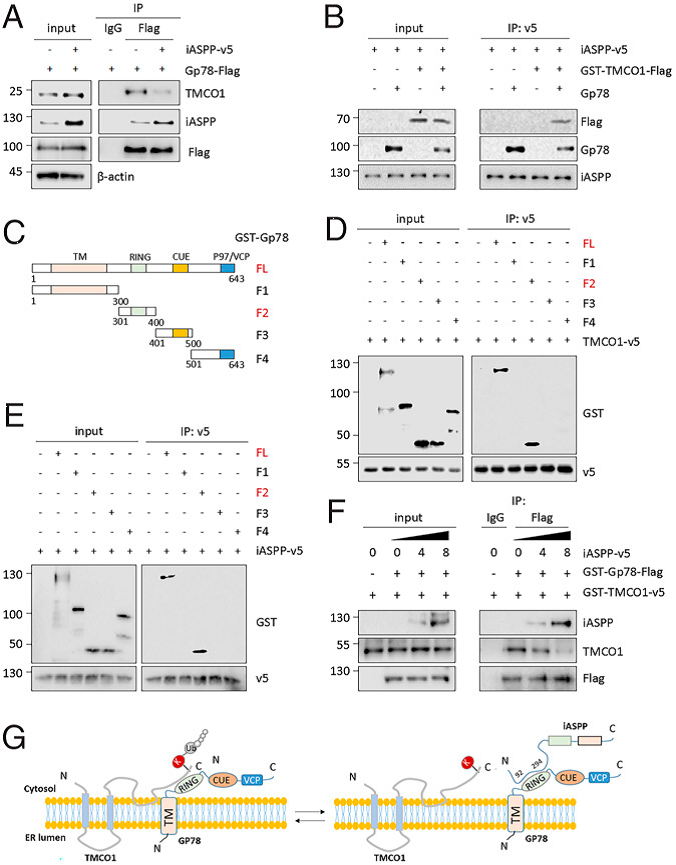
Fig 5 iASPP通过与Gp78竞争性结合稳定TMCO1
6、iASPP通过稳定TMC01 调节ER Ca2+的贮藏
鉴于TMCO1是一种Ca2+通道蛋白,iASPP对TMCO1具有调节作用,作者进一步研究了iASPP对Ca2+稳态的调节作用。为此,作者测量了HT-29和HCT-116细胞的Ca2+情况。游离Ca2+条件下存在离子霉素。离子霉素能导致Ca2+快速泄漏。因此检测到的胞质Ca2+通量间接反映了Ca2+含量。在这种条件下,用曲线下面积(AUC)定量的胞质Ca2+信号,值得注意的是,与相应的对照组相比,iASPP过表达显著降低了AUC(Figure 6A,B),而iASPP过表达显著提高了AUC(Figure 6C,D)。此外,iASPP对离子霉素诱导的Ca2+的影响随着TMCO1敲低完全消除。iASPP主要通过控制TMCO1蛋白水平来调控Ca2+稳态。

Fig 6 Ca2+通道蛋白TMCO1是iASPP调控ER Ca2+含量所必需的
7、ASPP-TMCO1在体内和体外均能促进肿瘤生长
TMCO1 敲低在软琼脂实验中显著减少HT-29和HCT-116细胞的集落形成,iASPP 敲低也起了类似的效果,而两者联合后没有进一步影响击落形成(Figure 7A)。iASPP-TMCO1对肿瘤生长的影响也通过裸鼠异种移植模型在体内进行了评估。结果发现,无论是iASPP还是TMCO1的敲低都能降低肿瘤生长和重量(Figure 7B,C)。不同移植瘤模型小鼠的体重无明显变化(Figure 7D)。与体外数据一致,作者发现iASPP 敲低导致异种移植瘤中TMCO1减少(Figure 7E)。此外,iASPP和TMCO1 敲低可增加细胞凋亡率,但同时敲低并不能进一步诱导细胞凋亡(Figure 7F)。
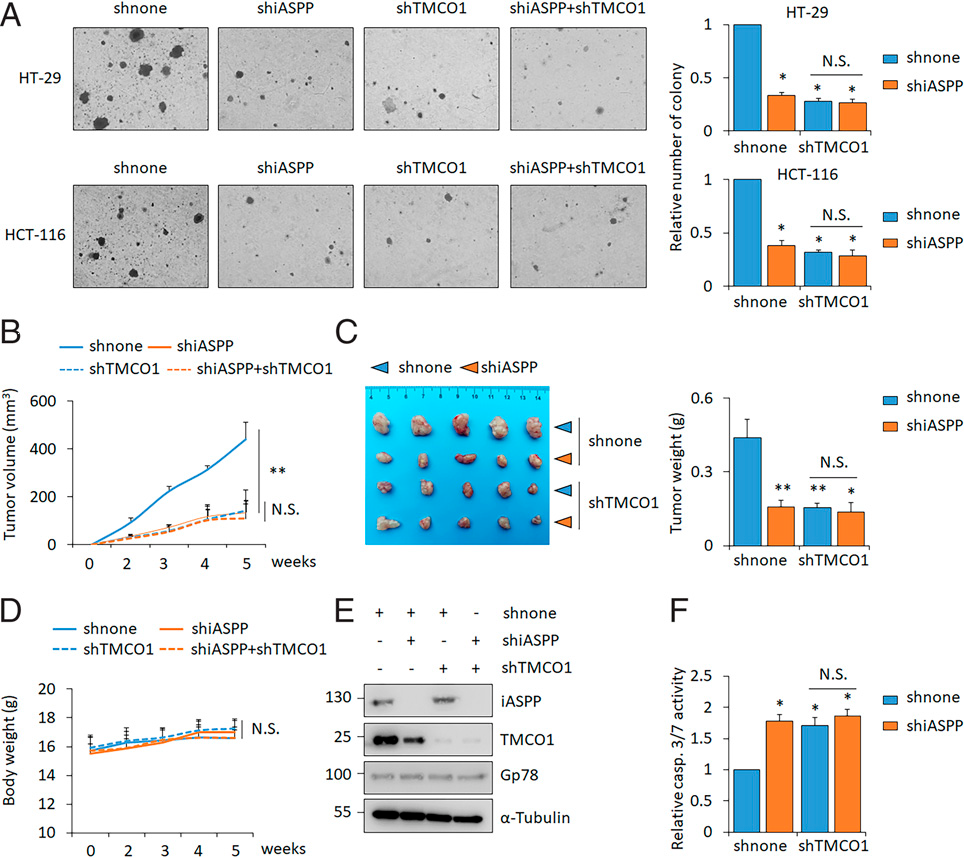
Fig 7 iASPP-TMCO1在体内和体外均能促进肿瘤生长
8、ASPP-TMCO1轴能降低凋亡Ca2+信号
用TG或组胺刺激内质网Ca2+释放,并检测对照组及iASPP或TMCO1敲低对细胞凋亡率的影响。结果发现,TG或组胺处理24 h后,凋亡水平(如caspase 3/7活性和 膜联蛋白v阳性率)增加(Figure 8A,B)。无论是在无应激条件下还是在TG或组胺处理条件下,iASPP或TMCO1敲低的细胞中, 膜联蛋白v凋亡细胞的百分比比对照组高得多(Figure 8A,B)。然而,在Ca2+非依赖性凋亡诱导剂依托泊苷的作用下,iASPP敲低促进了凋亡敏感性(Figure 8A,B)。iASPP敲低显著抑制了十字孢碱作用下HT-29细胞的集落形成。TMCO1敲低具有相似的效果,但是
两者同时敲低并没有进一步抑制集落形成(Figure 8C)。作者进一步验证了iASPP-TMCO1在HT-29异种移植模型小鼠体内的作用。十字孢碱抑制肿瘤生长,iASPP或TMCO1都能诱导HT29异种移植瘤对十字孢碱的反应。然而,两者同时敲低并没有产生额外的效应(Fig. 8D–F)。此外,在异种抑制瘤中,通过WB证实了iASPP敲低和TMCO1敲低的有效性。此外,iASPP敲低抑制了TMCO1的表达(Figure. 8G)。同样的,通过抑制iASPP或TMCO,十字孢碱诱导的凋亡增加,但两者同时敲低并没有进一步诱导凋亡(Figure. 8H)。这些数据共同表明,抑制iASPP-TMCO1轴使癌细胞对Ca2+调节的细胞死亡敏感。
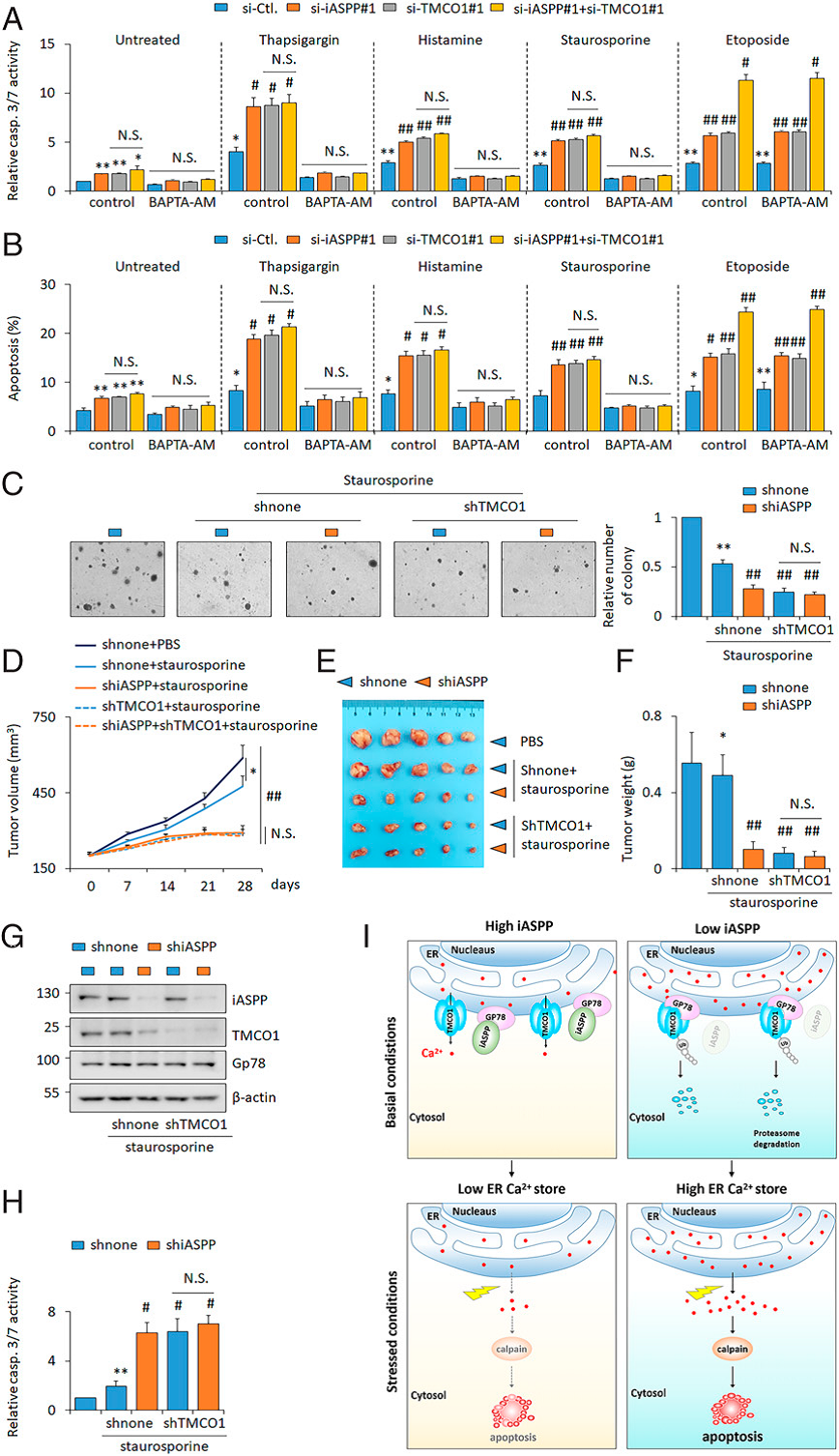
Fig 8抑制iASPP-TMCO1轴增加了在体内和体外十字孢碱敏感性
本研究阐明了iASPP调控Gp78介导TMCO1降解,导致Ca2+稳态失调,并增Ca2+引发的细胞凋亡的抗性。这些发现提示了能通过调节Ca2+稳态来治疗癌症。
参考文献:
Zheng, S., et al., iASPP suppresses Gp78-mediated TMCO1 degradation to maintain Ca(2+) homeostasis and control tumor growth and drug resistance. Proc Natl Acad Sci U S A, 2022. 119(6).