






去泛素酶USP27通过稳定SETD3促进细胞增殖和肝细胞癌进展
组蛋白甲基转移酶SETD3在各种生物事件中发挥着重要作用,其失调常与包括癌症在内的人类疾病有关。然而,潜在的监管机制仍然难以捉摸。近日,有文献报道了泛素特异性肽酶27 (USP27)通过与SETD3特异性相互作用,负向调节其泛素化作用,并增强其稳定性,从而促进肿瘤细胞的生长。该研究于2022年1月发表在《Cellular and Molecular Life Sciences》,IF:9.621。
技术路线:
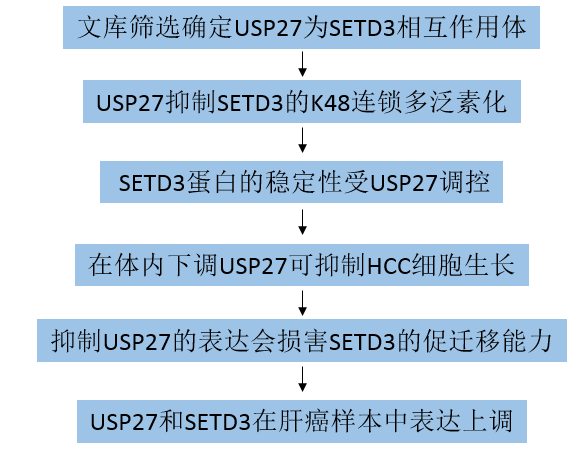
主要研究结果:
为了确定SETD3在肝脏肿瘤发生过程中异常表达的分子机制,利用DUB文库筛选SETD3相互作用的蛋白,该蛋白可能与SETD3的异常调控有关。通过免疫沉淀和western blot检测所示,SETD3被USP27拉下,SETD3也可以拉下USP27,说明USP27是真正的SETD3交互伙伴(图1A和B)。内源性SETD3是否与Hep3B细胞中的USP27相互作用:在anti-USP27中检测到SETD3,但在正常兔IgG免疫沉淀中未检测到(图1C)。此外,体外GST pull-down、PLA两个实验均证实了USP27与SETD3相互作用(图1D-F)。最后,通过突变体确定了USP27与SETD3之间的相互作用区域:USP27的C端UCH域(图1G-I)。此外,突变分析表明,SETD3与USP27的相互作用需要RSB域(图1J)。综上所述,这些数据表明USP27与SETD3相互作用。
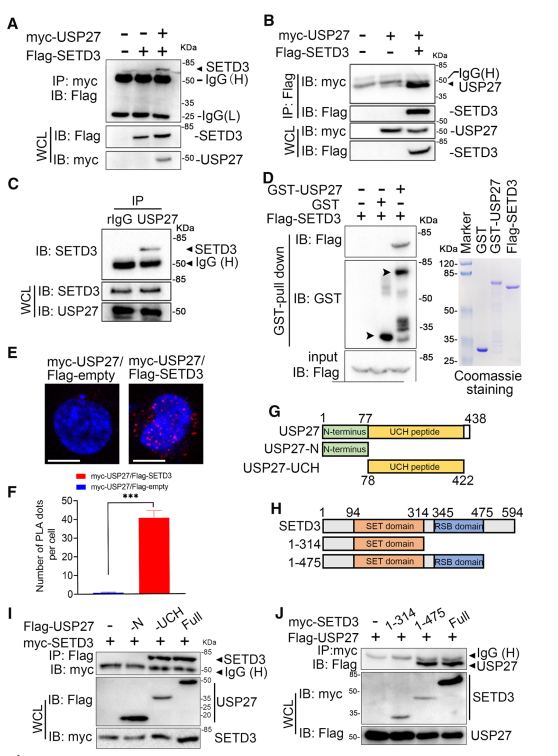
图1 USP27与SETD3相互作用
由于USP27是一种DUB(去泛素化化酶),可能会保护其底物免受蛋白酶体介导的降解,接下来确定该DUB是否会从SETD3中去除泛素。如图2A所示,在存在USP27表达的情况下,与SETD3连接的多聚素链被完全去除。与野生型(WT) USP27相比,USP27的酶活性突变体(C87A)未能消除SETD3蛋白的泛素化(图2B和C),这意味着USP27需要DUB活性才能从SETD3中去除泛素。USP27介导的SETD3去泛素化是非常特异性的,因为USP39并没有显著影响SETD3去泛素化(图2D)。此外,与shRNA对照相比,USP27表达的下调导致SETD3泛素化升高(图2E)。因此,为了确定SETD3上的多聚泛素链的哪一种泛素连锁是受USP27调控的,使用泛素突变体,其中只有位于11 (K11O)、48 (K48O)或63(K63O)位点的一个赖氨酸残基可被泛素化。如图2F所示,USP27过表达降低了SETD3的K48-连锁泛素化,但对SETD3的K11-或K63 -连锁泛素化无影响。总的来说,这些结果表明USP27是SETD3的一个真正的DUB,并以DUB活性依赖的方式去除SETD3的K48连锁多泛素化。
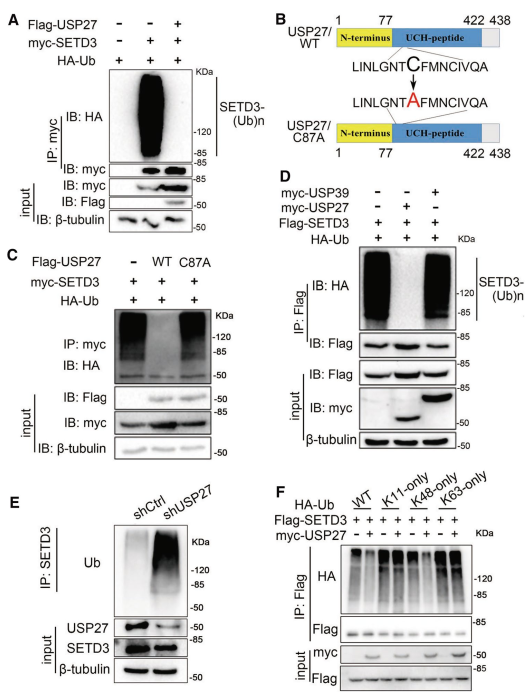
图2 USP27抑制SETD3的K48连锁多泛素化
3. SETD3蛋白的稳定性受USP27调控
之前研究发现USP27能够促进SETD3在K48位点的去泛素化。为了研究这种翻译后修饰是否促进了SETD3的稳定性,将SETD3表达构建物与空载体(EV)或USP27质粒共转染293 T细胞,检测SETD3在不同时间环己亚胺(CHX)作用下的蛋白水平。如图3A、B所示,USP27共转染显著上调SETD3蛋白表达,USP27显著延长SETD3蛋白的半衰期。此外,USP27的异位表达上调了内源性SETD3蛋白水平(图3C和D)。USP27/CA突变体的催化活性不能保护SETD3免受降解(图3E和F)。为了进一步证实USP27调节SETD3稳定性的观点,在Hep3B细胞中敲除了USP27的表达,发现内源性SETD3蛋白水平比对照组下降得更快(图3G和H)。此外,蛋白酶体特异性抑制剂MG132可以保护SETD3免受降解(图3I, J)。说明SETD3蛋白降解是通过蛋白酶体途径进行的。最后,在USP27过表达的Hep3B细胞中,SETD3 mRNA表达水平没有明显变化(图3K),提示USP27可能在转录后水平调控SETD3蛋白降解。综上所述,以上数据表明USP27可以正向调控SETD3蛋白的稳定性。
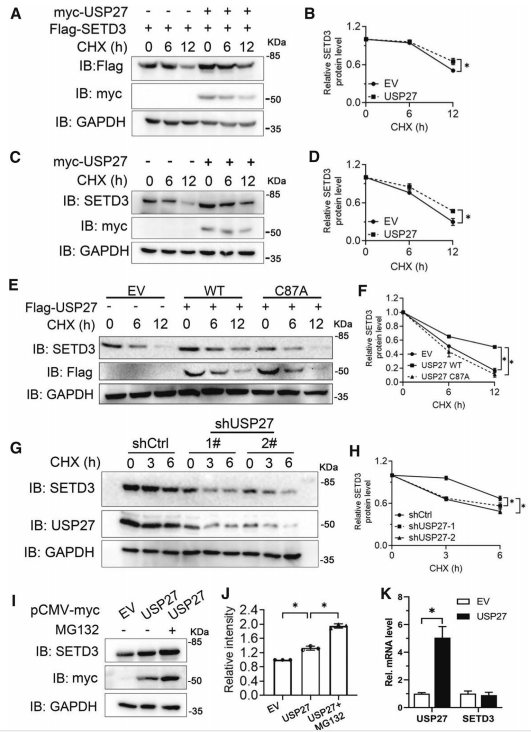
图3 USP27促进SETD3稳定
4. SETD3逆转USP27基因敲除介导的细胞增殖阻滞
由于USP27具有去泛素化和稳定SETD3的作用,可能通过调节SETD3蛋白水平来促进细胞增殖和迁移。为了验证这个假设,首先评估了生物USP27在肝癌中的作用 (图4)。如图4 A和B所示, 敲除USP27明显减少Hep3B和MHCC97H细胞的细胞生存能力。将SETD3引入USP27基因敲除细胞,可部分恢复其恶性表型,而这两种蛋白的敲除均可显著抑制细胞能力。集落形成实验进一步证实,USP27在Hep3B或MHCC97H细胞中的稳定敲除显著抑制了集落形成(图4C-F)。此外,5-EdU显示,USP27或SETD3的敲除与EdU标记细胞的显著下降百分率相关。在USP27或SETD3敲除细胞中加入SETD3的表达可以部分逆转(图4G和H)。同样,Ki67免疫荧光染色也证实了增殖下降,因为在USP27或SETD3敲除细胞中Ki67阳性增殖细胞的比例显著降低。Ki67阳性细胞的增加也提示更多的G0静止细胞在SETD3过表达后重新进入细胞周期,因为Ki67只在细胞周期的活动期而不是G0静止期表达。这些结果也表明,随着USP27或SETD3基因的下调,G0/G1期细胞部分的总大小增加,从而抑制细胞周期的进展。总的来说,这些发现表明USP27和SETD3对细胞增殖至关重要。
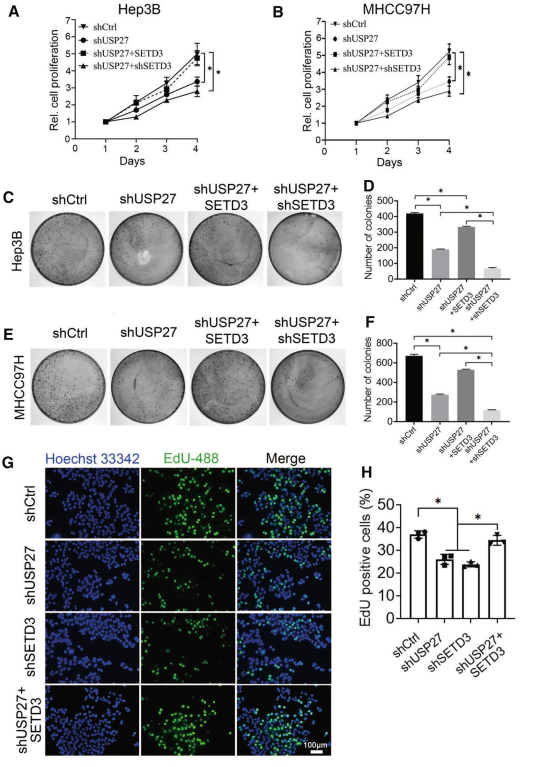
图4抑制USP27表达可阻断细胞增殖,延缓细胞生长
5. 在体内下调USP27可抑制HCC细胞生长
为了评价USP27和SETD3在体内对细胞生长的影响,将Hep3B细胞系皮下注射到裸鼠右侧侧腹建立异种移植模型。在肿瘤发展过程中,从注射后第6天开始每3天测量一次肿瘤体积,结果显示USP27和SETD3敲除细胞的肿瘤生长均显著下降(图5A)。注射后27天,解剖肿瘤,拍照(图5B)并称重(图5C)。USP27或SETD3敲除细胞产生的肿瘤明显比对照细胞更小、更轻,而在USP27缺失细胞中过表达SETD3可恢复肿瘤生长。此外,通过Ki67染色检测,USP27基因敲除细胞的肿瘤细胞活力也有所下降(图5D)。这些结果表明,USP27的缺失也抑制了肝癌细胞的体内生长。
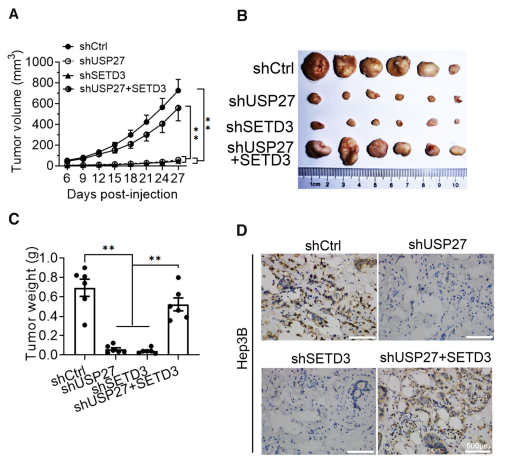
图5体内下调USP27基因可抑制HCC细胞的生长
6. 抑制USP27的表达会损害SETD3的促迁移能力
由于SETD3的上调可促进肝脏肿瘤发生和肿瘤进展,因此USP27也可能通过稳定SETD3来增强转移表型。为了探究USP27-SETD3轴在细胞迁移和侵袭中的作用,进行了创伤愈合实验,结果表明,与对照细胞相比,SETD3或USP27基因的下调显著抑制了细胞迁移。而将SETD3引入USP27基因敲低的细胞则部分恢复了转移表型(图6A-D)。Transwell侵袭实验一致显示,USP27和SETD3在Hep3B细胞(图6E, F)和MHCC97H细胞(图5G, H)的肿瘤侵袭中的作用类似。综上所述,这些数据表明,USP27或SETD3的下调可以阻止肝癌细胞的迁移和侵袭。
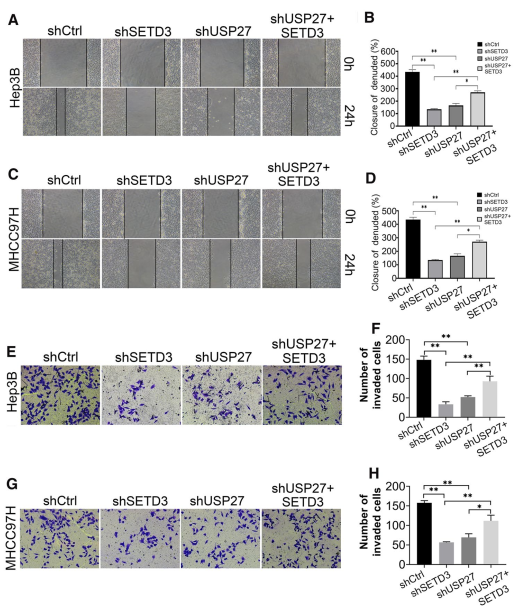
图6 抑制USP27表达可损害肝细胞癌细胞中SETD3的促迁移能力
7. USP27和SETD3在肝癌样本中表达上调
USP27-SETD3轴能够促进细胞增殖和迁移,推测它们可能在肝细胞癌中表达上调。首先,使用western blot检测了USP27和SETD3在HCC组织(n = 10)和匹配的相邻正常肝组织(n = 10)中的表达水平(图7A)。如图7B所示:USP27与SETD3蛋白水平明显上调且呈正相关。通过对肝癌组织和正常组织的免疫组化染色进一步检测两者之间的关系:与匹配的正常肝组织相比,SETD3和USP27在HCC组织中的表达水平显著上调(图7C)。根据患者的mRNA表达水平分为高表达组(高于正常组织平均水平)和低表达组(低于或等于正常组织平均水平),比较两组患者的生存期:与低表达的患者相比,USP27高表达患者的5年生存率明显降低(图7D)。SETD3的分析也得到了类似的结果。基于以上研究结果,作者得出结论,SETD3被USP27去泛素化,其蛋白水平受到USP27表达的正调控,并与USP27表达相关。肝细胞癌患者USP27的上调导致SETD3表达升高,进而促进细胞增殖、侵袭、迁移和肿瘤发生。

图7肝细胞癌组织中USP27和SETD3表达上调
主要结论:
在这里,该研究阐明了通过USP27的翻译后修饰对SETD3的动态调节(图8)。首先发现USP27是Hep3B细胞中SETD3的相互作用伙伴。USP27能够去泛素化并稳定SETD3。此外,体内外研究表明,USP27的缺失抑制了HCC细胞的增殖、侵袭、转移和肿瘤发生,而SETD3的过表达挽救了这种表型。SETD3和USP27水平在HCC中明显高于相关相邻组织。利用TCGA和GTEx数据库的数据,发现USP27和SETD3的表达水平与HCC患者的5年生存率呈负相关。总之,这项研究中的发现可以更好地理解USP27-SETD3轴在癌症发展中的作用和分子机制,为癌症治疗提供新的策略。
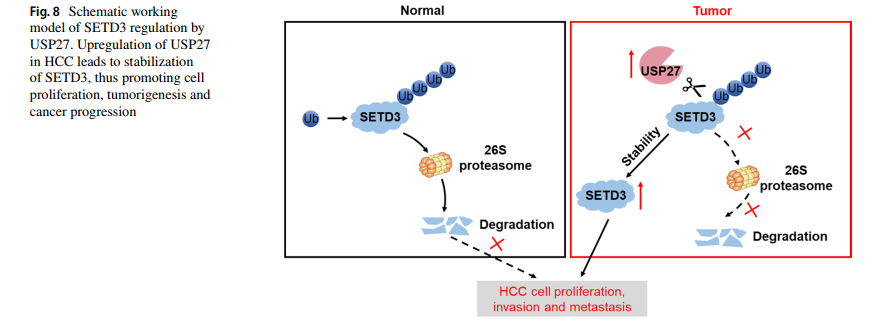
图8 USP27调节SETD3的模型示意图
参考文献:
Zou T, Wang Y, Dong L, Che T, Zhao H, Yan X, Lin Z. Stabilization of SETD3 by deubiquitinase USP27 enhances cell proliferation and hepatocellular carcinoma progression. Cell Mol Life Sci. 2022;79(1):70.