






肿瘤芽来源的CCL5招募成纤维细胞,通过CCR5-SLC25A24信号通路促进结直肠癌进展
肿瘤出芽在结直肠癌(CRC)的常规诊断中,被认为是独立于TNM分期的肿瘤预后因素。近日,有研究发现:在结直肠癌(CRC)的侵袭前沿,肿瘤芽源性趋化因子配体5(CCL5)可以通过CCR5 SLC25A24信号招募成纤维细胞,通过招募的成纤维细胞进一步促进血管生成和胶原合成,最终形成促肿瘤微环境(TME)。因此,CCL5可能成为CRC肿瘤出芽的潜在诊断标志物和治疗靶点。该研究发表在《Journal of Experimental & Clinical Cancer Research》,IF:11.161。
技术路线:
主要研究结果:
1. CRC肿瘤细胞可将成纤维细胞招募到肿瘤芽中
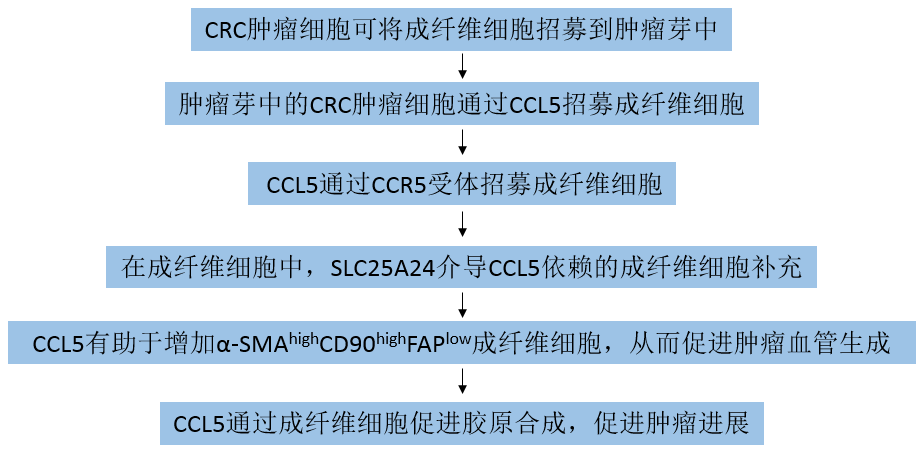
作者使用H&E染色对195个临床CRC组织样本进行评估,结果显示,CRC癌组织浸润前沿的肿瘤出芽比未出芽的其他肿瘤部位有更多的成纤维细胞(图1a)。肿瘤芽周围的成纤维细胞显示出α-SMA和CD90的高表达,但FAP的低表达(α-SMAhigh CD90 high FAPlow)(图1b)。这些结果表明,CRC组织浸润前沿的肿瘤出芽与成纤维细胞异质性密切相关。作者又通过检测上皮标记物E-cadherin、成纤维细胞标记物vimentin和α-SMA来鉴定成纤维细胞(图1c)。
通过共培养招募试验,发现FHC(人正常结直肠上皮细胞系)和LS174T、RKO、DLD-1、Caco2(人CRC细胞系)具有较弱的招募成纤维细胞的能力。相比之下,HCT-8、HCT116、HCT-15和SW620(人CRC细胞株)表现出更强的招募能力(图1d和e)。由于HCT116细胞在体外表现出强大的招募成纤维细胞的能力,被用于进一步的活体原位CRC异种移植小鼠实验。使用H&E染色检查小鼠盲肠中的肿瘤,观察到HCT116也可以在体内招募成纤维细胞(图1f)。综上所述,这些数据表明CRC的肿瘤芽可以招募成纤维细胞。
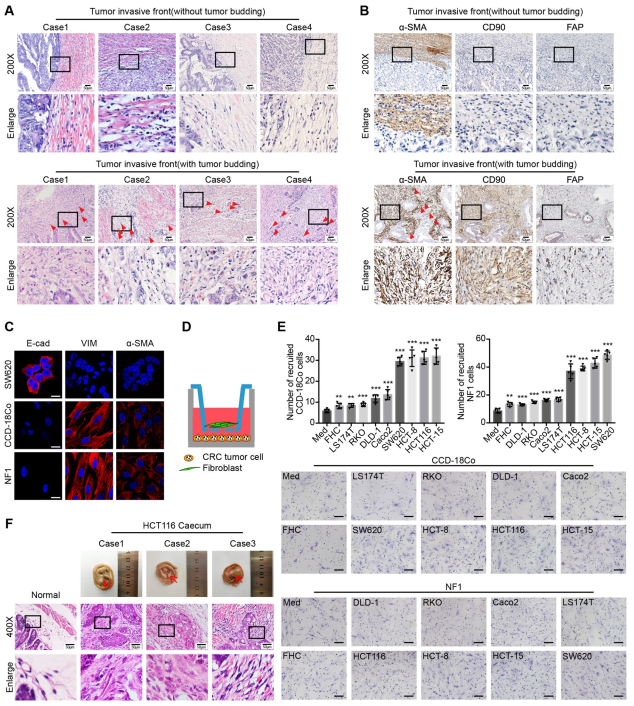
图1结直肠癌肿瘤细胞可将成纤维细胞招募到肿瘤芽中
2. 肿瘤芽中的CRC肿瘤细胞通过CCL5招募成纤维细胞
为了进一步确定结直肠癌肿瘤细胞分泌的关键细胞因子是导致成纤维细胞募集的原因,使用人类细胞因子阵列进行分析(图2a)。GO功能富集分析表明,差异表达的蛋白质参与各种活动(图2b)。这表明,CRC肿瘤细胞分泌的细胞因子可以招募TME中的其他细胞,并可能参与招募成纤维细胞,而成纤维细胞是细胞外基质的主要来源。
在差异表达的蛋白中,CCL5在人类CRC肿瘤细胞的CM样本中表达最多(图2c)。在FHC和8个人CRC肿瘤细胞株中检测CCL5的mRNA和分泌蛋白水平,结果与共培养招募试验的初步结果完全一致(图2d, e)。此外,CCD-18Co和人类原代正常结直肠成纤维细胞分泌的CCL5水平低于FHC(图2f)。因此,CCL5介导的成纤维细胞的招募似乎是通过旁分泌而不是自分泌信号发生的。接下来,用免疫组化法检测人CRC组织中CCL5的表达,结果显示CCL5在侵袭前部的肿瘤芽中高表达(图2g)。为了检验CCL5是否为成纤维细胞招募的关键细胞因子,再次进行招募实验(图2h)。CCL5可以招募到CCD-18Co和不同的人类原代正常结直肠成纤维细胞(图2i)。最后,为了确定招募成纤维细胞的CCL5是否由CRC肿瘤细胞分泌,分别用siRNA和慢病毒构建了CCL5敲除和过表达的细胞系。共培养招募实验(图2j)显示,敲除CCL5后,肿瘤细胞招募成纤维细胞的能力明显减弱(图2k)。相比之下,当CCL5过表达时,这种能力增强(图2l)。招募分析,CM样本稳定细胞(图2m)显示效果类似于观察整个细胞(图2 n, o)。这些结果表明CCL5在CRC肿瘤芽中高度表达,并能从TME招募成纤维细胞。

图2肿瘤芽中的CRC肿瘤细胞通过CCL5募集成纤维细胞
3. CCL5通过CCR5受体招募成纤维细胞
为了探究CCL5是否通过它的受体介导成纤维细胞的招募,使用siRNA分别下调它们在成纤维细胞中的表达,并验证了干扰效率(图3a, b)。接下来,进行招募检测,如图3c所示:只有在成纤维细胞中下调CCR5才能显著减弱CCL5招募成纤维细胞的能力(图3d)。
此外,在共培养招募试验之前和期间,分别使用CCR1抑制剂BX471和CCR5抑制剂Maraviroc治疗成纤维细胞,并阻断CCR1和CCR5(图3e)。只有Maraviroc治疗显著削弱了CCL5招募成纤维细胞的能力(图3f)。以上结果强烈表明CCL5通过CCR5受体参与成纤维细胞招募。
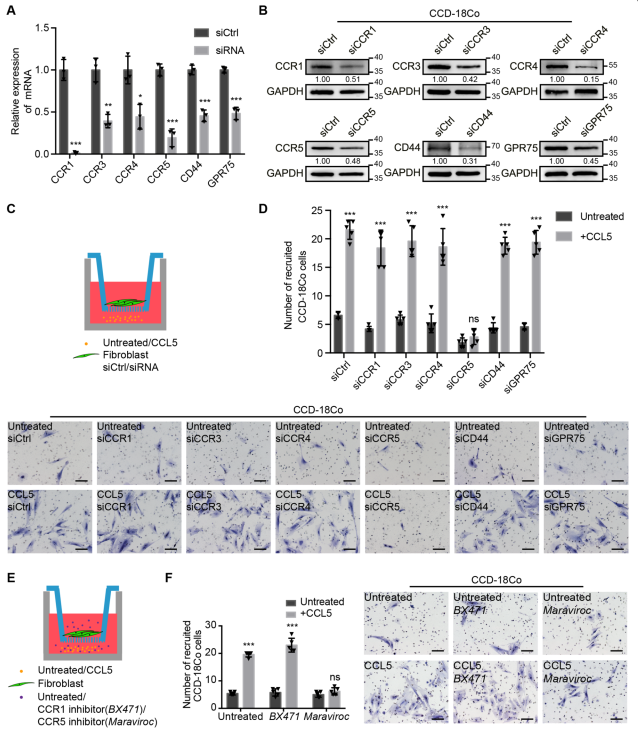
图3 CCL5通过CCR5受体招募成纤维细胞
4. 在成纤维细胞中,SLC25A24介导CCL5依赖的成纤维细胞补充
为了探究CCL5刺激后成纤维细胞的胞内变化,使用CCL5治疗前后的成纤维细胞进行了转录组测序(图4a)。选取差异表达前20个基因进行验证:在CCL5治疗后,SLC25A24 mRNA水平在人类原代正常结直肠成纤维细胞中上调(图4b)。
采用IF法检测SLC25A24蛋白在人结直肠癌组织中的表达和定位。结果显示,SLC25A24在侵袭前部肿瘤芽周围的成纤维细胞中高度表达(图4c)。在体内,在sh-CCL5转染的CRC细胞的肿瘤异种移植中,观察到成纤维细胞中SLC25A24的表达减少(图4d)。
功能上,为了探索CCL5介导的成纤维细胞招募是否依赖于SLC25A24,将抗SLC25A24的siRNA转染成纤维细胞,并进行招募实验(图4e)。成纤维细胞中SLC25A24表达的减少显著削弱了CCL5招募成纤维细胞的能力(图4f, g)。这表明CCL5介导的成纤维细胞招募依赖于SLC25A24和CCR5。
KEGG富集分析表示PI3K-Akt信号通路在CCL5刺激后最为活跃(图4h)。免疫印迹结果显示,CCL5可以促进成纤维细胞Akt和mTOR的磷酸化(图4i,左图)。相比之下,当SLC25A24的表达被抑制时,CCL5刺激后成纤维细胞磷酸化的Akt和mTOR水平下降(图4i,右图)。这些发现表明CCL5通过成纤维细胞中的SLC25A24- pAkt-pmTOR轴招募成纤维细胞。
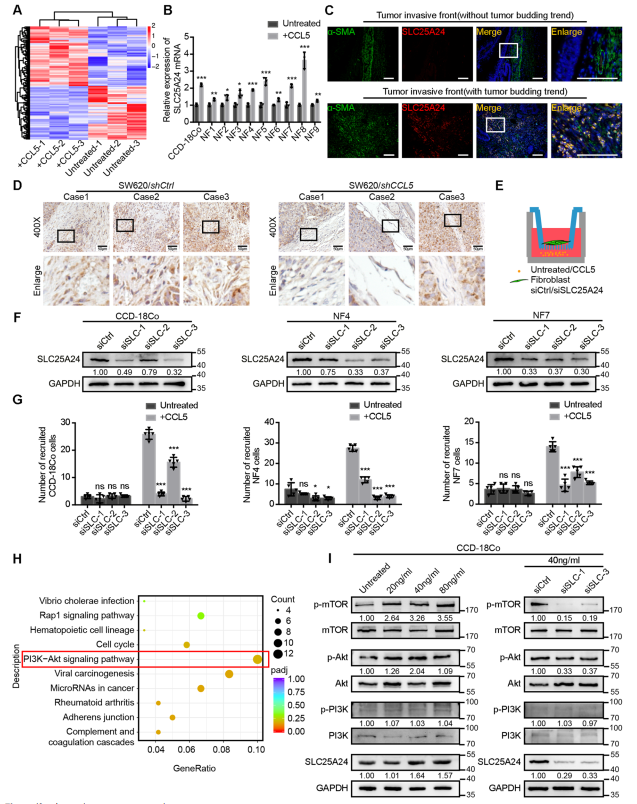
图4成纤维细胞中SLC25A24介导CCL5依赖性成纤维细胞补充
5. CCL5有助于增加α-SMAhigh CD90high FAPlow成纤维细胞,从而促进肿瘤血管生成
最初发现(图1b)证明肿瘤芽周围的成纤维细胞是α-SMAhigh, CD90high和FAPlow。为了检验CCL5是否是增加α-SMAhigh CD90high FAPlow成纤维细胞的主要原因,进行了IF试验。该实验表明,经CCL5处理的成纤维细胞中α-SMA和CD90的表达升高(图5a)。下一步研究CCL5在成纤维细胞介导的血管生成中的作用。免疫印迹显示CCL5处理后成纤维细胞血管内皮标志物FLI1、VE-cadherin、CD31、VEGFA水平以及α-SMA和CD90水平升高(图5b)。流式细胞术也显示,在CCL5刺激后,VE-cadherin+成纤维细胞亚群的比例增加(图5c)。CCL5刺激后增强成纤维细胞的促血管生成能力(图5d)。在侵袭性前沿,CCL5在肿瘤芽中高表达,肿瘤芽被大量的α-SMAhigh CD90high FAPlow成纤维细胞和血管包围(图5e)。综上,肿瘤芽源CCL5增加了α-SMAhigh CD90high FAPlow成纤维细胞的数量,并通过增加VEGFA和成纤维细胞转分化为血管内皮细胞来促进肿瘤血管生成。
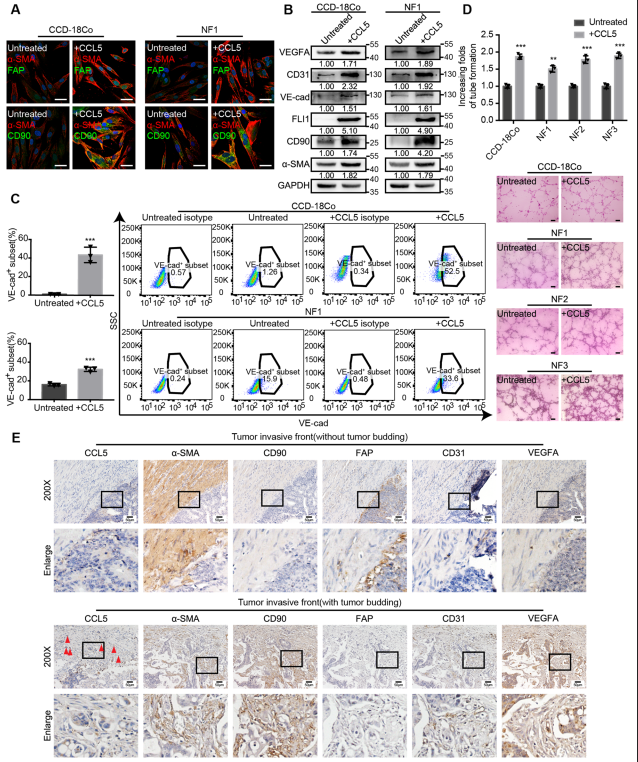
图5 CCL5有助于增加α-SMAhigh CD90high FAPlow成纤维细胞,从而促进肿瘤血管生成
6. CCL5通过成纤维细胞促进胶原合成,促进肿瘤进展
GO功能富集分析表明,CCL5也参与细胞外基质相关功能(图6a)。WB分析显示CCL5可以增加体外成纤维细胞中COL1和COL3的蛋白表达(图6b)。临床样本中,CRC组织中CCL5的表达明显高于正常组织(图6c)。采用Pearson’sχ2检验分析CCL5表达与临床特征之间的相关性(表1)。进一步的Spearman相关性试验表明,CCL5高水平表达与肿瘤芽增加的高风险(图6 d)呈正相关。此外,对195例人结直肠癌组织标本中的88例进行了COL1和COL3染色,以检测胶原分布。结果显示结肠结直肠癌组织中COL1和COL3表达升高(图6e)。采用Pearson’sχ2检验分析Sirius Red染色与CRC临床特征的相关性(表2)。进一步的Spearman相关性检验显示,CRC肿瘤细胞周围胶原蛋白的高分布与肿瘤芽的增加呈正相关(图6f)。CCL5在侵袭性前缘肿瘤芽中的高表达往往伴随着胶原合成的增加(图6g)。Spearman相关性分析显示,同一CRC组织样本中,肿瘤细胞中CCL5的表达水平与周围胶原蛋白的数量呈正相关(图6h)。
随后,利用分泌大量CCL5的HCT116细胞建立原位CRC异种移植小鼠模型进行体内分析。发现侵袭性前沿的肿瘤细胞周围胶原合成增加(图6i)。利用人CRC基因芯片图谱GSE39582分析COL1或COL3低表达和高表达患者的无复发生存期:COL1和COL3的高表达与CRC患者预后不良密切相关(图6j)。综上所述,肿瘤芽分泌CCL5,然后通过成纤维细胞促进胶原合成,从而促进肿瘤进展。
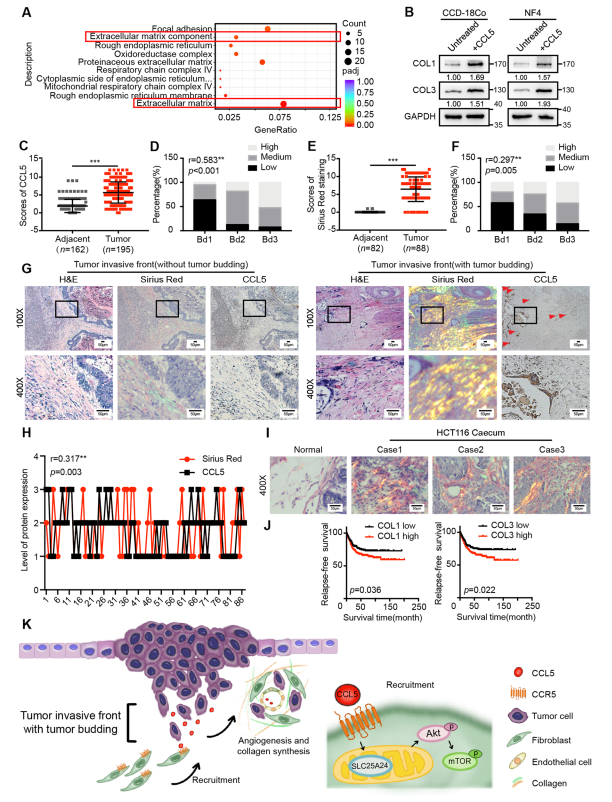
图6 CCL5通过成纤维细胞促进胶原合成,促进肿瘤进展
主要结论:
本研究发现CRC肿瘤芽分泌高水平的CCL5,CCL5通过CCR5- SLC25A24信号征集成纤维细胞,导致侵袭前沿肿瘤芽周围形成特征性成纤维细胞簇。这进一步促进了肿瘤血管生成和胶原合成,促进了恶性进展(图6k)。因此,该研究为CCL5可能作为CRC肿瘤出芽的潜在诊断标志物和治疗靶点提供了证据。
参考文献:
Gao LF, Zhong Y, Long T, Wang X, Zhu JX, Wang XY, Hu ZY, Li ZG. Tumor bud-derived CCL5 recruits fibroblasts and promotes colorectal cancer progression via CCR5-SLC25A24 signaling. J Exp Clin Cancer Res. 2022; 41:81.