






S6K1介导的PDK1磷酸化受损削弱AKT激酶活性和致癌作用
蛋白质激酶作为细胞信号转导的中心节点,在时间和空间上能使下游底物发生磷酸化,以催化传递来自细胞外或细胞内变化的信号。3-磷酸肌醇依赖蛋白激酶1 (PDK1)作为一种主激酶,在磷酸化和激活蛋白激酶A、B和C (AGC)家族激酶(包括AKT)中起着重要作用,但目前对PDK1的上游调控作用少有报道。本研究报道了核糖体蛋白S6激酶β1 (S6K1)直接磷酸化蛋白同源结构域,并破坏PDK1与AKT的相互作用和活化。本文于2022年3月发表于 《Nature Communications》,IF=14.919。
本文技术路线:

本文主要内容:
作者推测上游激酶可能介导PDK1-S549磷酸化,于是分析S549周围氨基酸情况,发现S549位点内存在一个经典的AGC激酶磷酸化共识基序位点(RxRxxS/T) (Fig 1a)。为了评估AGC激酶家族是否可以促进PDK1在该位点的磷酸化,筛选了一组AGC激酶,包括AKT1 (myr-AKT1),AKT2 (myr-AKT2),S6K1 (S6K1- r3a)和SGK1 (SGK1-Δ60)。用内源性的AKT底物磷酸化抗体检测到S6K1,而不是其他的AGC激酶在内源性水平上显著增强PDK1磷酸化(Fig 1b)。胰岛素诱导的PDK1磷酸化可被mTORC1或S6K抑制剂显著拮抗,AKT抑制剂的拮抗程度较低,同时S6K下游底物核糖体蛋白S6的磷酸化事件减少(Fig 1c)。与此一致的是,S6K1可以显著增强PDK1的磷酸化,且呈剂量依赖性(Fig 1d),而S6K1诱导的PDK1磷酸化可被mTORC1或S6K抑制剂减弱 (Fig 1e)。进一步观察发现PDK1磷酸化随胰岛素刺激而波动。此外,胰岛素在S6K1和S6K2双敲除(S6K1 /2−/−)小鼠胚胎的成纤维细胞(mef)中极大促进了PDK1磷酸化(Fig 1f)。提示S6K1在胰岛素诱导中可能是必需的基因磷酸化。与这一发现一致的是,癌细胞中S6K1的缺失显著减少了PDK1的磷酸化 (Fig 1g)。
为了验证S549是否确实是PDK1上主要的S6K1磷酸化残基,作者发现S549A敲除降低了S6K1介导的PDK1磷酸化 (Fig 1h)。与这一发现一致的是,在体外S6K1可以直接磷酸化WT突变体,而不能磷酸化S549A突变体 (Fig 1i)。为了进一步检测S549位点的PDK1磷酸化,作者构建并验证了磷酸化抗体特异性识别的PDK1-pS549,并观察到S6K1确实可以在细胞的S549位点磷酸化PDK1 (Fig 1b-h),也能被mTORC1或S6K抑制剂可以抑制 (Fig 1c)。这些结果表明S6K1能直接磷酸化PDK1的Ser549残基。
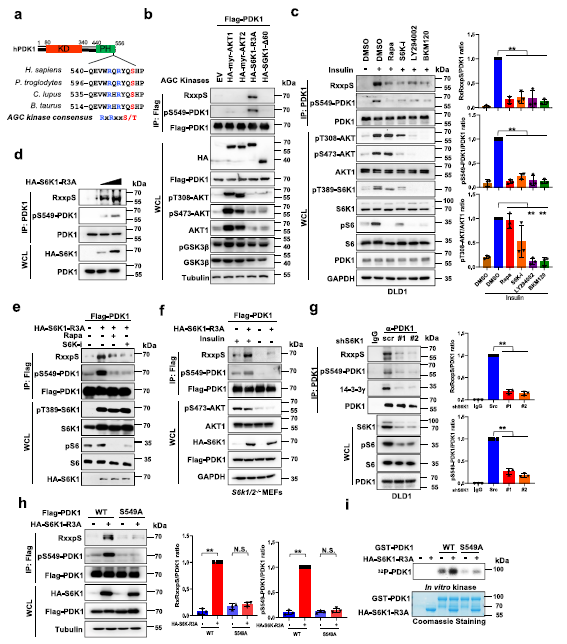
Fig1 S6K直接使PDK1在Ser549位点磷酸化
2 S6K1介导的PDK1磷酸化抑制AKT激酶活性和致癌功能
为了探究S6K1介导的PDK1磷酸化的生物学功能,作者将PDK1的不同突变形式(WT、S549A和S549D)引入DLD1-PDK1−/−细胞中,发现与WT或PDK1-S549D相比、PDK1-S549A表达显著升高AKT-pT308 (Fig 2a)。此外,在胰岛素处理条件下,如pGSK3β,与表达PDK1-WT-或S549D的细胞相比,DLD1-PDK1−/−表达PDK1-S549A的细胞中pT308-AKT及其下游底物显著升高(Fig 2b,c)。
作者发现,与WT或S549D突变体PDK1相比,引入缺磷型PDK1 (S549A)可以显著促进细胞集落形成和锚定独立生长 (Fig 2d,e)。为了揭示PDK1-S549磷酸化在体内对肿瘤生长的作用,作者利用异种移植小鼠模型,发现与表达细胞S549的DLD1-PDK1−/−的细胞相比,PDK1-S549A的表达明显促进DLD1-PDK1−/−细胞肿瘤的生长 (Fig 2f–j)。同时,与PDK1-WT或S549D的表达的肿瘤相比较,携带PDK1-S549A的肿瘤表现出更多的Ki67染色,同时AKT下游底物pS6增加 (Fig 2k,l)。这些结果表明,在体内和体外,S6K介导的PDK1磷酸化负调控AKT激酶活性和致癌功能。
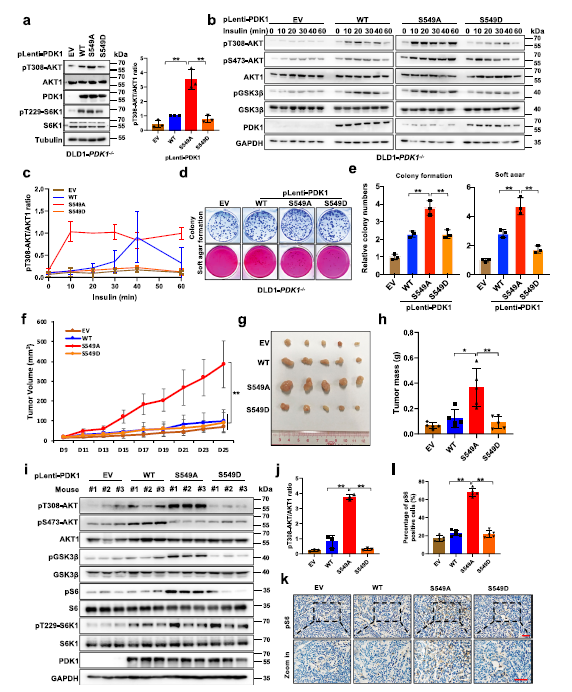
Fig2 PDK1的磷酸化抑制AKT激酶的活性和致癌功能
3 PDK1- S549磷酸化显著减弱PDK1与PIP3的相互作用和质膜定位
为了研究S6K1介导的PDK1磷酸化是否影响其激酶活性,作者对PDK1进行了体外激酶测定。结果表明,S6K1-R3A的组成活性形式与PDK1- wt、S549A或S549D变体仅轻度影响PDK1磷酸化其底物AKT1的能力 (Fig 3a),表明S6K1介导的PDK1磷酸化对AKT激酶活性的抑制可能不是由于PDK1酶活性降低所致。接下来,作者发现PDK1和S6K1-R3A可显著降低PDK1与AKT的相互作用,且呈剂量依赖性,且S549处PDK1的磷酸化水平增加 (Fig 3b)。通过磷酸化IRS1/2来排除S6K已建立的负反馈作用的影响,观察PDK1-S549D或S549A与AKT之间的互作,结果发现PDK1-S549A的相互作用增强,而S549D减弱,但不影响与其他PDK1底物(如SGK1、PLK1或S6K1)的相互作用 (Fig 3c)。
已有研究表明PDK1和AKT的相互作用主要发生在PIP3定位的质膜上,PIP3的pull-down检测结果显示,S6K1-R3A显著降低了WT的相互作用,而PDK1的S549A突变体与PIP3的相互作用没有降低(Fig 3d)。同时,抑制S6K1活性可以促进PDK1与PIP3的相互作用(Fig 3e)。这些数据表明,S549磷酸化可以使PDK1从质膜和PIP3中分离出来,从而降低PDK1与AKT的相互作用。与这一发现一致,作者观察到S6K1确实可以通过细胞分馏分析降低PDK1的膜定位或免疫染色 (Fig 3g)。因此,与WT或S549D形式的PDK1相比,S549A突变体增强了PDK1膜定位。
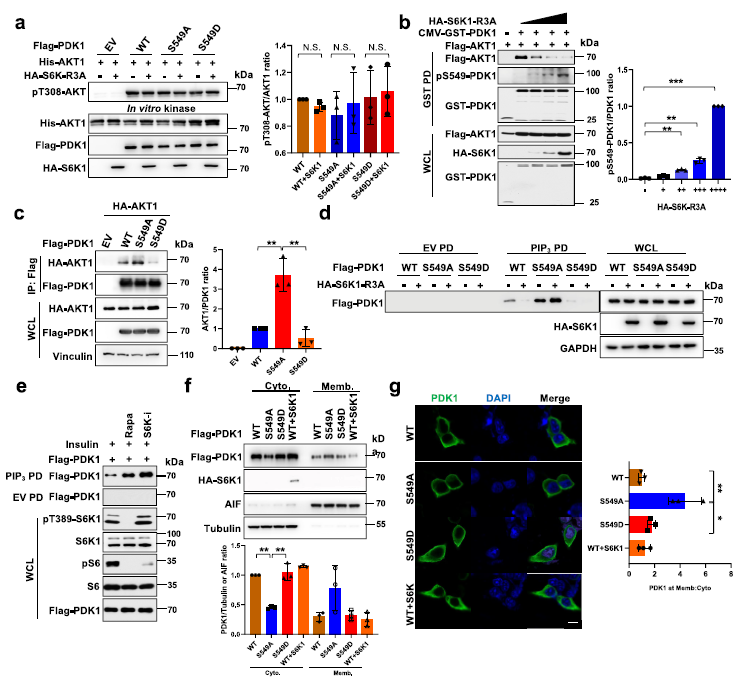
Fig 3 S6K1介导的PDK1磷酸化抑制PDK1膜定位和PIP3结合
4、14-3-3介导PDK1从PIP3中分离,并促进其二聚
为了揭示磷酸化PDK1与PIP3解离并形成二聚体的潜在机制,作者假设存在能够识别磷酸化PDK1的接头蛋白,并进一步干扰其与PIP3的相互作用。已有研究证实14-3-3接头蛋白在蛋白室转位中起主要作用,尤其是磷酸化蛋白。因此,作者对14-3-3家族成员进行了筛选,发现14-3-3γ可以在外源和内源水平上特异性地与PDK1相互作用 (Fig 4a–c)。值得注意的是,RxxpSxP这个基序与S6K1介导的PDK1磷酸化残基(Ser549)重叠,并且在不同物种之间进化保守 (Fig 4d)。有趣的是,与PDK1、AKT1、PLK1和SIN1衍生的不同PH结构域相比,作者观察到只有PDK1-PH参与14-3-3γ的结合。更重要的是,S6K1- r3a显著增强,而S6K1的消耗减少了PDK1与14-3-3γ之间的互作 (Fig 4g)。在细胞和体外实验中,S549D突变体PDK1增强,而S549A突变体PDK1减弱PDK1与14-3-3γ的相互作用 (Fig 4e,f)。
先前有报道称,磷酸化丝氨酸或苏氨酸后的脯氨酸残基对14-3-3相互作用至关重要。因此,无论在细胞内还是体外,PDK1- p551a突变均能强烈减弱PDK1与14-3-3γ的相互作用。然而,P551A突变体并没有消除S6K介导的PDK1与PIP3的分离 (Fig 4g),从而增加了AKT的相互作用,这不能被异位表达S6K1-R3A破坏(Fig 4g)。 S6K1-R3A或PDK1- S549d的异位表达同样增强了14-3-3γ向PDK1的招募(Fig 4g)。此外,P551A突变体增强了其与PIP3的结合,S6K1-R3A的表达不会干扰PIP3的结合(Fig 4i)。
通过凝胶过滤的方法,作者观察到PDK1和pS549-PDK1在其二聚体大小在150KD左右时,与14-3-3γ共聚在相似的部分(Fig 4j),表明14-3-3γ可能是磷酸化PDK1进行二聚反应。此外,消耗14-3-3γ会破坏PDK1的二聚反应(Fig 4k),并增加PDK1与AKT的相互作用。更重要的是,14-3-3γ敲除显著提高了AKT及其下游靶点磷酸化水平(Fig 4l,m)。表明接头蛋白14-3-3γ在S6K1介导的AKT被PDK1抑制过程中起重要作用。这些研究结果表明,14-3-3γ以S6K1介导的S549磷酸化依赖的方式与PDK1结合,使PDK1与PIP3分离,进而促进PDK1二聚化,从而抑制AKT激酶活性(Fig 4n)。
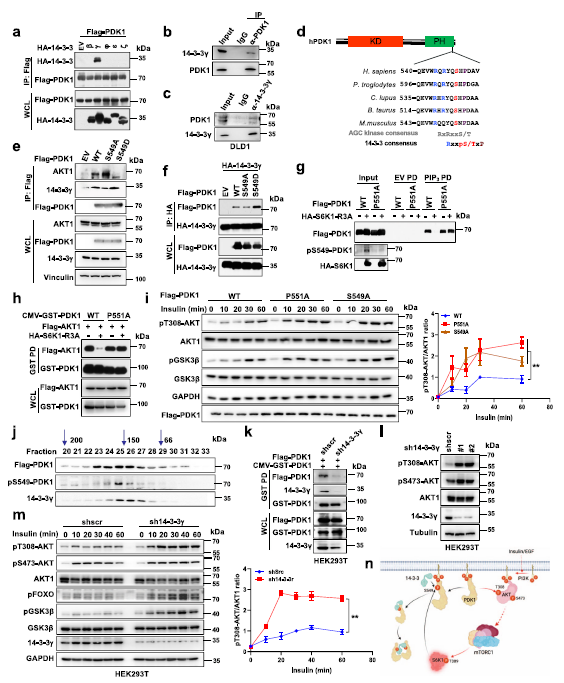
Fig4 14-3-3γ结合磷酸化的PDK1并将其从细胞质膜上解离
5.患者相关的PDK1突变削弱其与14-3-3的相互作用,促进AKT激酶活性和致癌功能
为探讨S6K1介导PDK1磷酸化在肿瘤发生中的病理作用,对癌症基因组数据进行分析,结果发现患者相关的PDK1突变发生在S6K1介导的PDK1磷酸化或14-3-3γ结合区域附近 (Fig 5a)。
接下来,这些突变被分成两组,I型突变位于可能阻断S6K1介导的磷酸化的区域; (R544K, R546P和S549N);II型突变(P551A, P551L和P551Q)可能在直接干扰对14-3-3γ的识别(Fig 5a)。结果发现, I型突变以及程度较轻II型突变,显著降低了S6K1介导的PDK1磷酸化(Fig 5b)。I型和II突变体显著减弱PDK1/14-3-3γ相互作用(Fig 5c),从而增加了PDK1与PIP3的结合 (Fig 5d),以及它们的膜定位(Fig 5e–h)。值得注意的是,这些突变增强了PDK1与AKT的相互作用(Fig 5i,j),同时增加了HEK293-sgPDK1细胞中AKT及其靶蛋白的磷酸化水平 (Fig 5k,l)。
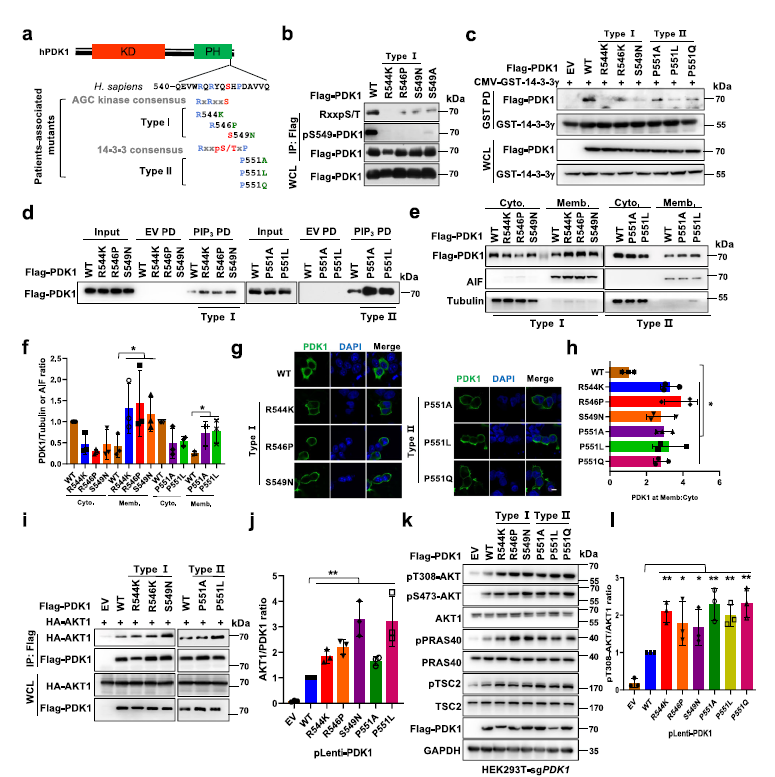
Fig5 患者相关的PDK1突变与PDK1膜位置和AKT激酶激活有关
此外,将这些变量重新引入DLD1-PDK1−/−或SW480-shP DK1细胞中增加了pT308-AKT的表达 (Fig 6a),并促进了癌细胞的致癌能力(Fig 6b,c)。此外,与表达WT-PDK1的细胞相比,这些突变体还促进了异种移植小鼠模型中肿瘤的生长,并增加了AKT下游的pS6,以及增殖标志物Ki67的表达(Fig 6d–g)。
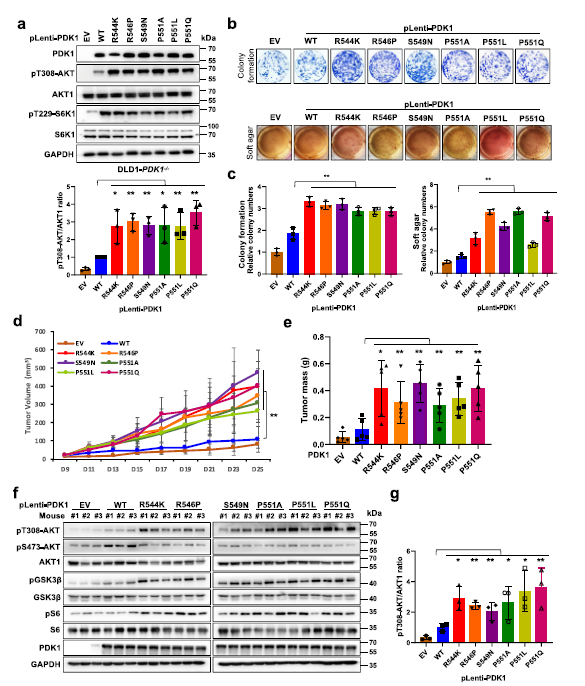
Fig 6患者来源的PDK1突变促进AKT1的激活和致癌功能
总之,S6K1直接磷酸化PDK1的PH结构域,增强PDK1结合14-3-3的能力,进而解离PIP3和质膜中的PDK1,导致PDK1/AKT相互作用减少,AKTT308磷酸化受损。这一观察结果表明,S6K1以不同的负反馈方式和磷酸化依赖方式紧密控制AKT激酶的活性(Fig 7)。

Fig 7 生理和病理条件下S6K1对PDK1负调控的模型
参考文献:
Jiang, Q., et al., S6K1-mediated phosphorylation of PDK1 impairs AKT kinase activity and oncogenic functions. Nat Commun, 2022. 13(1): p. 1548.