






胶质母细胞瘤抗血管生成治疗的靶点-小胶质细胞外泌体circKIF18A
多形性胶质母细胞瘤(GBM)是最致命的原发性肿瘤。研究GBM血管生成的新分子机制非常重要。研究发现,M2小胶质细胞极化与GBM患者微血管密度呈正相关。M2-胶质瘤相关的小胶质细胞(GAM)可通过将外泌体circKIF18A转运到hBMECs中促进GBM的血管生成。在机制上,circKIF18A可以结合hBMECs中FOXC2,维持其稳定性和核易位。此外,作为一种转录因子,FOXC2可以直接与ITGB3、CXCR4和DLL4的启动子结合并上调其表达。此外,FOXC2还可以激活PI3K/AKT信号,促进GBM的血管生成。我们的研究确定了M2-GAM衍生外泌体circKIF18A通过靶向FOXC2参与GBM血管生成的新分子机制。这可能为改善GBM抗血管生成治疗的疗效提供一个新的治疗靶点。本文于2022年5月发表于Oncogene(IF=8.756)上。
技术路线

结果
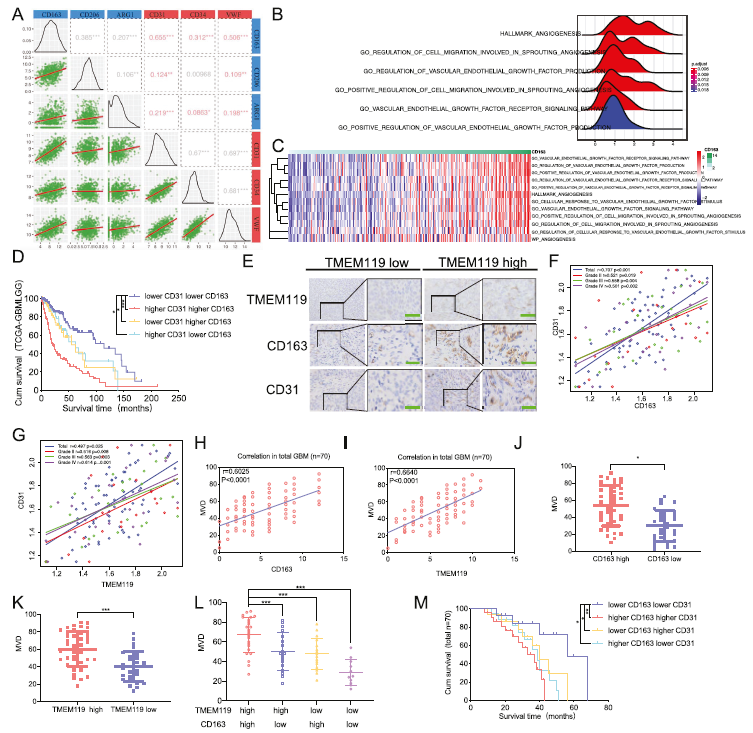
1)M2小胶质细胞与GBM患者微血管密度相关
首先,我们分析了TCGA、CGGA、Rembrandt和Gravendel数据库,发现M2极化标记基因(CD163、CD206、ARG1)和血管生成相关基因(CD31、CD34、vWF)之间存在正相关(图1A)。基于TCGA和CGGA数据库的GSEA和GSVA分析表明,CD163高表达组富含血管生成过程和信号传导(图1B、C)。此外,Kaplan-Meier生存分析表明,在TCGA、Rembrandt和Gravendel数据库中,CD163和CD31高表达的患者明显短于CD163和CD31低表达的患者(图1D)。随后,我们采用qPCR、免疫组化等方法检测70例胶质瘤患者中TMEM119、CD163、CD31的表达(图1E)。皮尔逊相关分析显示,CD163或TMEM119与CD31或微血管密度(MVD)之间存在很强的正相关关系(图1F-I)。值得注意的是,CD163和TMEM119高分组的MVD高于低分组(图1J-L)。Kaplan-Meier生存分析还表明,CD163和CD31表达较高的胶质瘤患者的中位生存时间短于CD163和CD31表达较低的胶质瘤患者(图1M)。总之,这些结果表明,M2小胶质细胞与GBM患者微血管密度呈正相关。
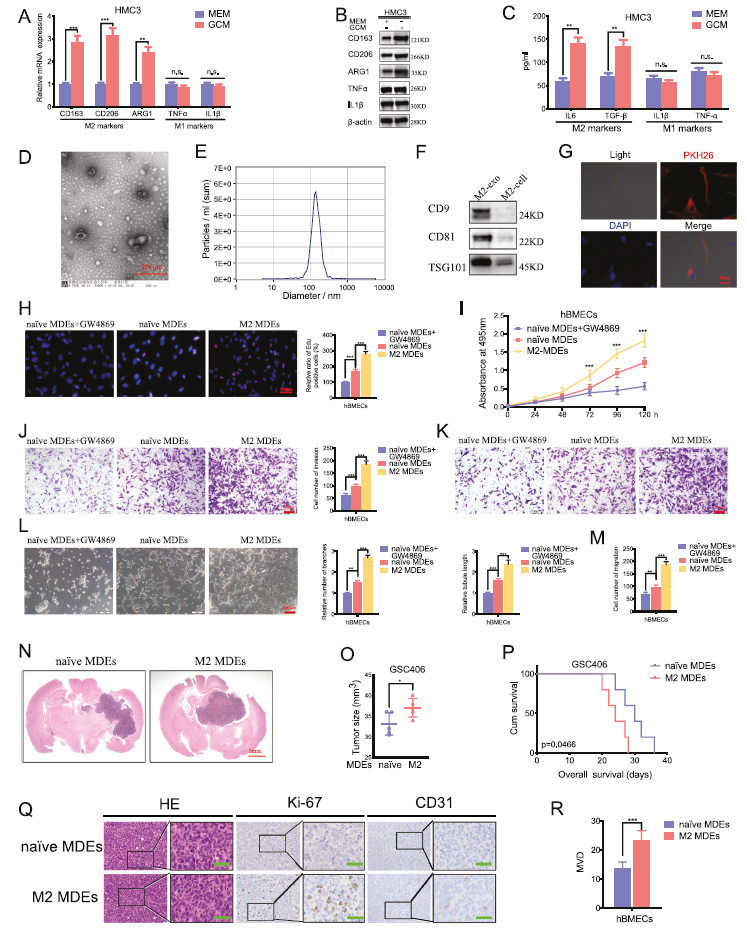
2)M2小胶质源性外泌体在体外和体内促进血管生成
我们通过qPCR、western blotting和ELISA分析验证原始HMC3的M2极化,M2标记物(CD163、CD206、ARG1、IL-6和TGF-β)显著上调,M1标记物(IL1β和TNF-α)下调(图2A-C)。我们分离纯化小胶质细胞来源外泌体(MDE),并通过透射电镜(TEM)观察(图2D)。NTA显示,室温下MDE的平均直径为132 nm(图2E)。通过western blotting检测MDE的标记基因(CD9、CD81和TSG101)(图2F)。此外,PKH26分析表明,HUVEC可以吸收MDEs(图2G)。我们进一步检测了M2-MDEs对hBMECs的直接影响。所有EDU、MTS、transwell、迁移和试管形成分析表明,与GW4869治疗组和原始MDEs治疗的组相比,M2- MDEs治疗后hBMECs的生存能力、侵袭、迁移和试管形成能力都有所提高(图2H-M)。总之,这些结果表明M2-MDEs在体外可以促进血管生成。
为了进一步证实M2-MDEs能够促进体内血管生成,我们构建了基于C57BL/6小鼠的同源GBM模型。我们将BV-2 M2或幼稚外泌体注射到GBM荷瘤小鼠体内。结果表明,M2-MDEs治疗后的肿瘤体积增大(图2N,O)。Kaplan-Meier生存分析表明,M2-MDEs治疗可比原始MDEs减少中位生存时间(图2P)。免疫组织化学显示,M2-MDEs治疗可以上调Ki67和CD31的表达以及MVD(图2Q,R)。因此,这些结果表明M2-MDEs在体内可促进GBM的肿瘤发生和血管生成。
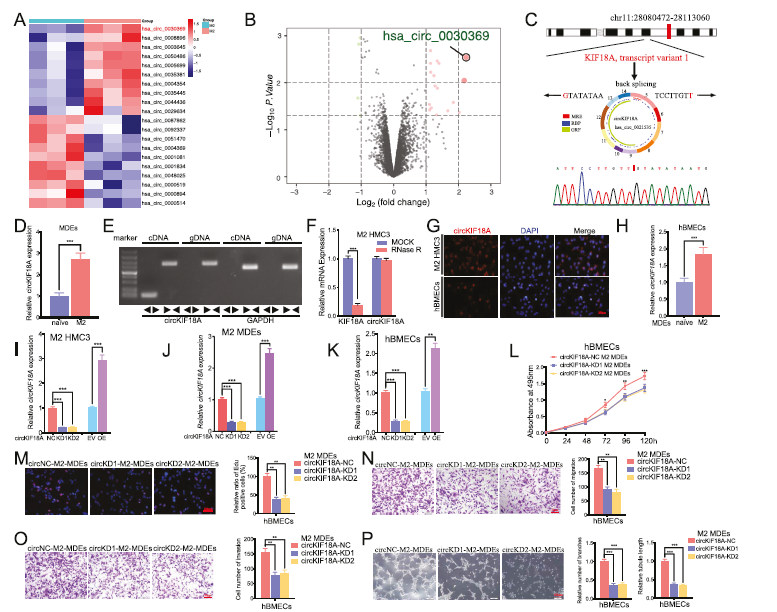
3)M2-MDEs将circKIF18A运输到hBMECs中
在HMC3的naïve和M2-MDEs之间进行CircRNA测序。结果显示,有17个差异表达的circRNAs。其中,与幼稚MDEs相比,circKIF18A是M2-MDEs中上调最高的CircRNA(图3A、B、D)。CircKIF18A是从KIF18A mRNA的转录本之一反向拼接而成,由其5-14个外显子组成。Sanger测序证实了头-尾剪接(图3C)。琼脂糖凝胶电泳显示,cDNA中的发散引物扩增了circKIF18A,但gDNA没有扩增(图3E)。此外,RNase R处理显示circKIF18A的表达略有变化,而其线性RNAs KIF18A明显减少(图3F)。FISH分析显示,circKIF18A主要位于M2 HCM3和hBMECs的细胞质中(图3G)。此外,qPCR用于检测MDEs治疗后hBMECs中circKIF18A的表达,结果显示,M2-MDEs治疗后circKIF18A的表达高于幼稚MDEs治疗的hBMECs(图3H)。然后,M2-HMC3中circKIF18A的表达被敲低或过表达,并通过qPCR进行验证(图3I)。同样,在circKIF18A-KD M2-MDEs治疗后,qPCR也显示circKIF18A-KD M2-MDEs(图3J)和hBMECs(图3K)中circKIF18A表达下调。然而,circKIF18A-OE M2-MDEs治疗后的结果相反(图3J,K)。总之,这些结果表明circKIF18A在M2-MDEs中过表达,并可通过MDEs转运到内皮细胞中。MTS、EDU、transwell、迁移和试管形成分析表明,与阴性对照(NC)M2-MDEs相比,circKIF18A-KD M2-MDEs处理后,hBMECs的细胞活力、增殖、侵袭、迁移、分支数量和小管长度均减少(图3L-P),提示M2小胶质细胞外泌体circKIF18A在体外可促进血管生成。
4)M2小胶质细胞外泌体circKIF18A可直接与FOXC2结合
由于M2极化与GBM患者微血管密度相关,我们基于CD163在TCGA和CGGA数据库中的表达进行了GSEA。我们收集了至少在两条血管生成途径中起作用的基因,发现了6个重叠基因,包括PTGS2、FLT4、SULF1、FOXC2、NRP1和VEGFA(图4A,B)。我们进一步探讨了circKIF18A与hBMECs中这些基因之间可能的分子机制。qPCR显示,在hBMECs中,经circKIF18A-KD或circKIF18A- OE M2-MDEs治疗后,这六个基因的mRNA表达没有持续变化(图4C)。然而,WB显示,在circKIF18A-KD M2-MDEs处理后,VEGFA和FOXC2表达下调,而在circKIF18A-OE M2-MDEs处理后,PTGS2、NRP1、FOXC2的蛋白表达上调(图4D)。因此,FOXC2可能是受circKIF18A调控的候选基因。根据catRAPID数据库的预测,circKIF18A可以与FOXC2蛋白结合,尤其是circKIF18A的Δ291–350区域与FOXC2蛋白的Δ376–427区域之间的相互作用位点(图4E)。RIP分析显示,抗FOXC2组中circKIF18A的相对富集度明显高于IgG治疗组。此外,在circKIF18A-OE M2-MDEs处理后,circKIF18A的富集程度更高,而在circKIF18A-KD M2-MDEs处理后,circKIF18A的富集程度降低(图4F)。RNA下拉分析还表明,生物素化的circKIF18A wt探针可以下拉hBMEsC中的FOXC2,而circKIF18A mt探针不能(图4G)。此外,将FOXC2 DNA片段克隆到pCMV5质粒中并转染到hBMECs中。RIP分析进一步显示,在pCMV5-Δ376–427组中,circKIF18A的富集程度更高(图4H)。这些结果表明,circKIF18A可以直接与FOXC2蛋白结合。MG-132处理hBMECs后,由circKIF18A-KD M2-MDE引起的FOXC2蛋白表达减少得到恢复(图4I)。CHX分析表明,在hBMECs中,经circKIF18A-KD M2-MDEs处理后,FOXC2蛋白的半衰期缩短,经circKIF18A-OE M2-MDEs处理后,FOXC2蛋白的半衰期延长(图4K,L)。此外,在hBMECs中,经circKIF18A-KD M2-MDEs处理后,FOXC2主要存在于细胞质中,几乎没有核分布,而经circKIF18A-OE M2-MDEs处理后,其核分布明显增加(图4M)。CircKIF18A-KD M2-MDEs处理下调了细胞核中FOXC2的表达,而CircKIF18A- OE M2-MDEs处理上调了细胞核中FOXC2的表达(图4J)。因此,这些结果表明M2-HMC3通过外泌体circKIF18A诱导FOXC2稳定性和核易位。

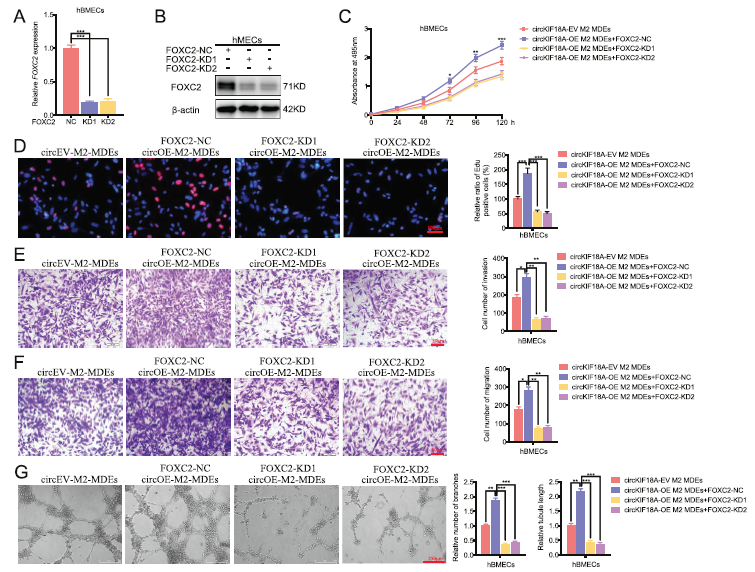
5)M2小胶质细胞外泌体circKIF18A通过FOXC2促进体外血管生成
为了进一步证实M2小胶质细胞外泌体circKIF18A通过靶向FOXC2促进血管生成,hBMECs中FOXC2的表达被基于慢病毒的RNAi沉默,并且qPCR和WB验证了这些效应(图5A,B)。尽管使用circKIF18A-OE M2-MDEs处理,MTS、EDU、transwell、migration和tube formation实验显示FOXC2敲除后,与阴性对照组相比,EDU阳性hBMECs的比率、hBMECs的侵袭和迁移数量、hBMECs的分支数量和小管长度均下降(图5C–G)。因此,这些结果表明,M2小胶质细胞外泌体circKIF18A在体外通过FOXC2促进血管生成。
6)M2小胶质外泌体circKIF18A可促进FOXC2对ITGB3、CXCR4、DLL4和PI3K/AKT信号通路的转录调控能力
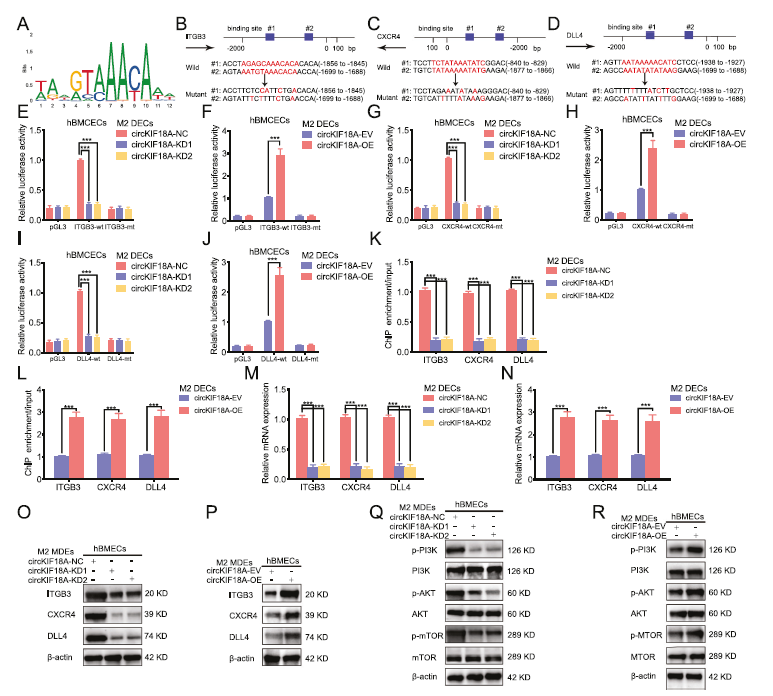
我们根据Jaspar数据库的预测进一步进行了荧光素酶报告分析和芯片分析(图6A-D)。结果表明,在circKIF18A-KD M2-MDEs治疗后,pGL3-ITGB3-wt、pGL3-CXCR4-wt和pGL3-DLL4-wt的相对荧光素酶活性均下调,而在circKIF18A-OE M2-MDEs治疗后,其相对荧光素酶活性增强(图6E–J)。芯片分析还表明,在使用circKIF18A-KD M2-MDEs处理后,抗FOXC2可降低hBMECs中ITGB3、CXCR4和DLL4的富集度(图6K),而在使用circKIF18A-OE M2-MDEs处理后获得相反的结果(图6L)。此外,qPCR和WB也证实了circKIF18A-KD或circKIF18A-OE M2-MDEs处理后ITGB3、CXCR4和DLL4的表达水平(图6M-P)。据报道,FOXC2可以通过激活PI3K/AKT信号促进肿瘤的恶性表型。用WB检测PI3K/AKT信号的活性。结果表明,在hBMECs中,经circKIF18A-KD MDEs治疗后,p-PI3K、p-AKT和p-MTOR的表达均下调(图6Q),而经circKIF18A-OE MDEs治疗后,结果相反(图6R)。
7)M2小胶质细胞外泌体circKIF18A促进体内肿瘤生长和血管生成
Mmu_U circ_0009168是小鼠体内circKIF18A的同源circRNA。其在BV-2中的过表达和敲除通过qPCR进行验证(图3E)。然后将mmu_circ_0009168-KD或mmu_circ_0009168-OE M2-MDEs注射到C57BL/6小鼠的同源GBM模型中。结果表明,与NC M2-MDEs相比,mmu_circ_0009168-OE M2-MDEs治疗后肿瘤体积明显增大(图7A,B)。Kaplan-Meier生存分析表明,mmu_circ_0009168-OE M2-MDEs比NC M2-MDEs能缩短中位生存时间(图7C)。免疫组织化学显示,mmu_circ_0009168-OE M2-MDEs治疗后,Ki-67、CD31和MVD的表达增加(图7D,H)。然而,mmu_circ_0009168-KD M2-MDEs治疗后的结果相反(图7D-H)。这些结果表明,M2小胶质细胞外泌体circKIF18A在体内促进肿瘤的生长和血管生成。为了说明我们的发现,图7I中的示意图显示,胶质母细胞瘤相关的小胶质细胞来源的外泌体circKIF18A通过靶向FOXC2促进血管生成。
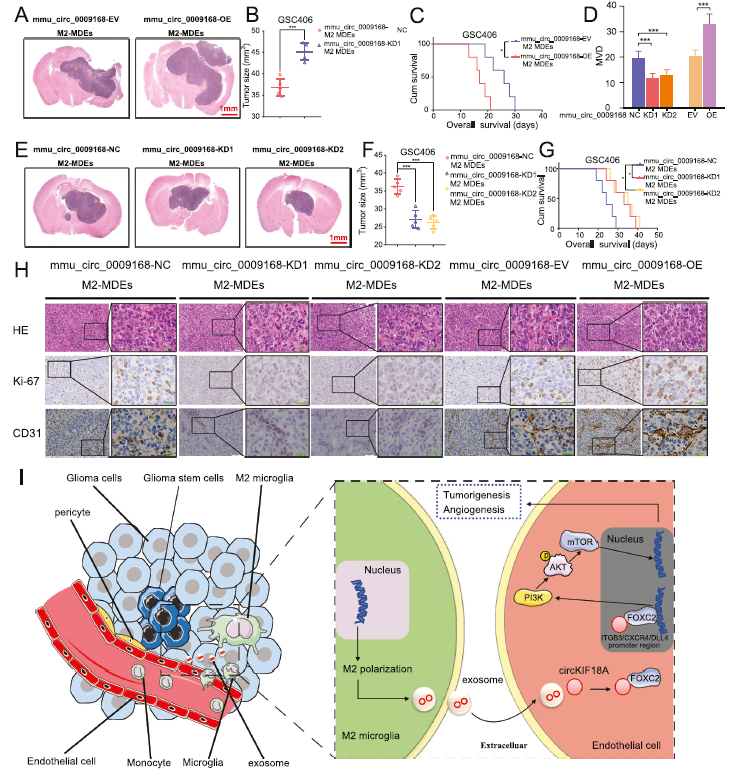
结论:M2小胶质细胞通过运输外泌体circKIF18A进入 hBMECs,促进GBM血管生成。从机制上讲,circKIF18A可以结合hBMECs中FOXC2,维持其稳定性和核转位。FOXC2可以通过转录调控ITGB3、CXCR4、DLL4的表达,激活PI3K/AKT信号通路。我们的研究为GBM的抗血管生成治疗提供了新的机制和靶点,这可能会改善抗血管生成治疗的效果。
参考文献:
Jiang Y, Zhao J, Xu J, Zhang H, Zhou J, Li H, Zhang G, Xu K, Jing Z. Glioblastoma-associated microglia-derived exosomal circKIF18A promotes angiogenesis by targeting FOXC2. Oncogene. 2022;41(26):3461-3473. doi: 10.1038/s41388-022-02360-4.