






MiRNA-363-3p/DUSP10/JNK轴参与弥漫大B细胞淋巴瘤化疗耐药
蒽环类化疗耐药是弥漫大B细胞淋巴瘤(DLBCL)面临的主要挑战。我们观察并验证了miRNA-363-3p的高表达与化疗耐药性之间的独立相关性。体外和体内实验表明,MiRNA-363-3p可通过DLBCL细胞系中的异位表达和CRISPR/Cas9介导的敲除减少阿霉素诱导的凋亡和肿瘤缩小。我们还发现DNA甲基化参与了miRNA-363-3p的转录调控。进一步研究表明,DUSP10是miRNA-363-3p的靶点,其抑制可促进JNK磷酸化。miRNA-363-3p/DUSP10/JNK轴主要与同源重组(HR)和DNA修复途径的负调控相关。miRNA-363-3p的异位表达可更有效地修复阿霉素诱导的双链断裂(DSB),同时增强非同源末端连接修复,减少HR修复。靶向JNK和聚ADP核糖聚合酶1可显著抑制阿霉素诱导的DSB修复,增加阿霉素诱导的细胞凋亡和肿瘤缩小,提高荷瘤小鼠的生存率。总之,miRNA-363-3p/DUSP10/JNK轴是DLBCL中一种新的耐药机制,可通过靶向治疗逆转。本文于2022年3月发表于Leukemia(IF=11.528)杂志上。
技术路线

结果
1)高水平的miRNA-363-3p与蒽环类化疗耐药显著相关
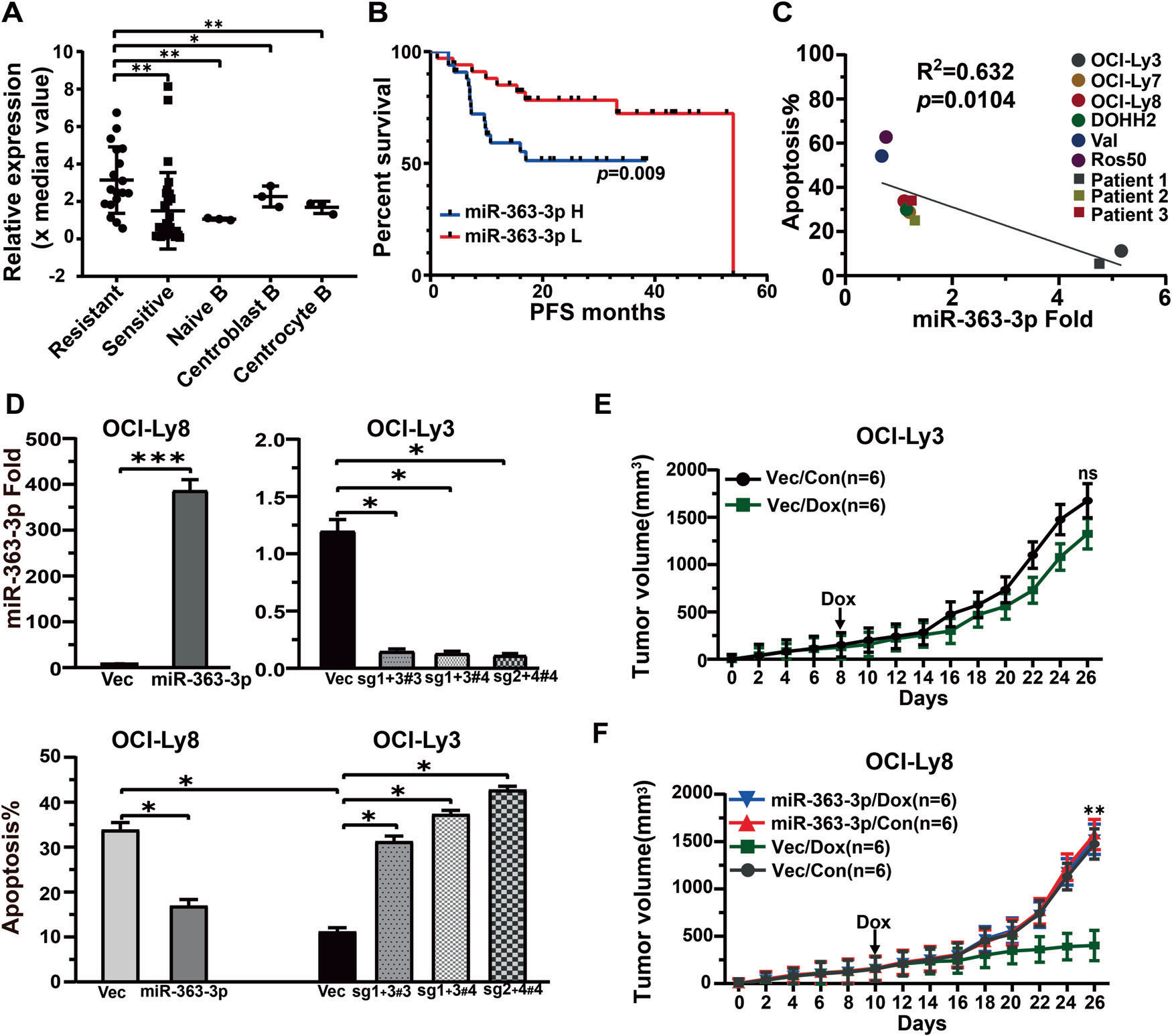
对47例DLBCL患者冷冻保存的肿瘤标本进行MiRNA表达谱分析。耐药组的miRNA-363-3p水平显著升高(图1A)。miRNA-363-3p水平高的患者表现出不利的PFS(图1B)。在B细胞淋巴瘤细胞系中也观察到miRNA-363-3p水平的高差异。在治疗48小时后,miRNA-363-3p水平与阿霉素(25 ng/ml)诱导的6株DLBCL细胞和3株DLBCL患者肿瘤细胞凋亡呈负相关(图1C)。在miRNA-363-3p异位的OCI-Ly8中观察到阿霉素诱导的凋亡显著减少,而在CRISPR/cas9介导的miRNA-363-3p敲除的OCI-Ly3中检测到凋亡显著增加(图1D)。体内实验证实了miRNA-363-3p表达与阿霉素耐药之间的关系。与OCI-Ly3载体相比,阿霉素诱导小鼠肿瘤明显缩小,而miRNA-363-3p的异位表达显著降低了阿霉素对OCI-Ly8的作用(图1E, F)。
2)MiRNA-363-3p受DNA甲基化的转录调控
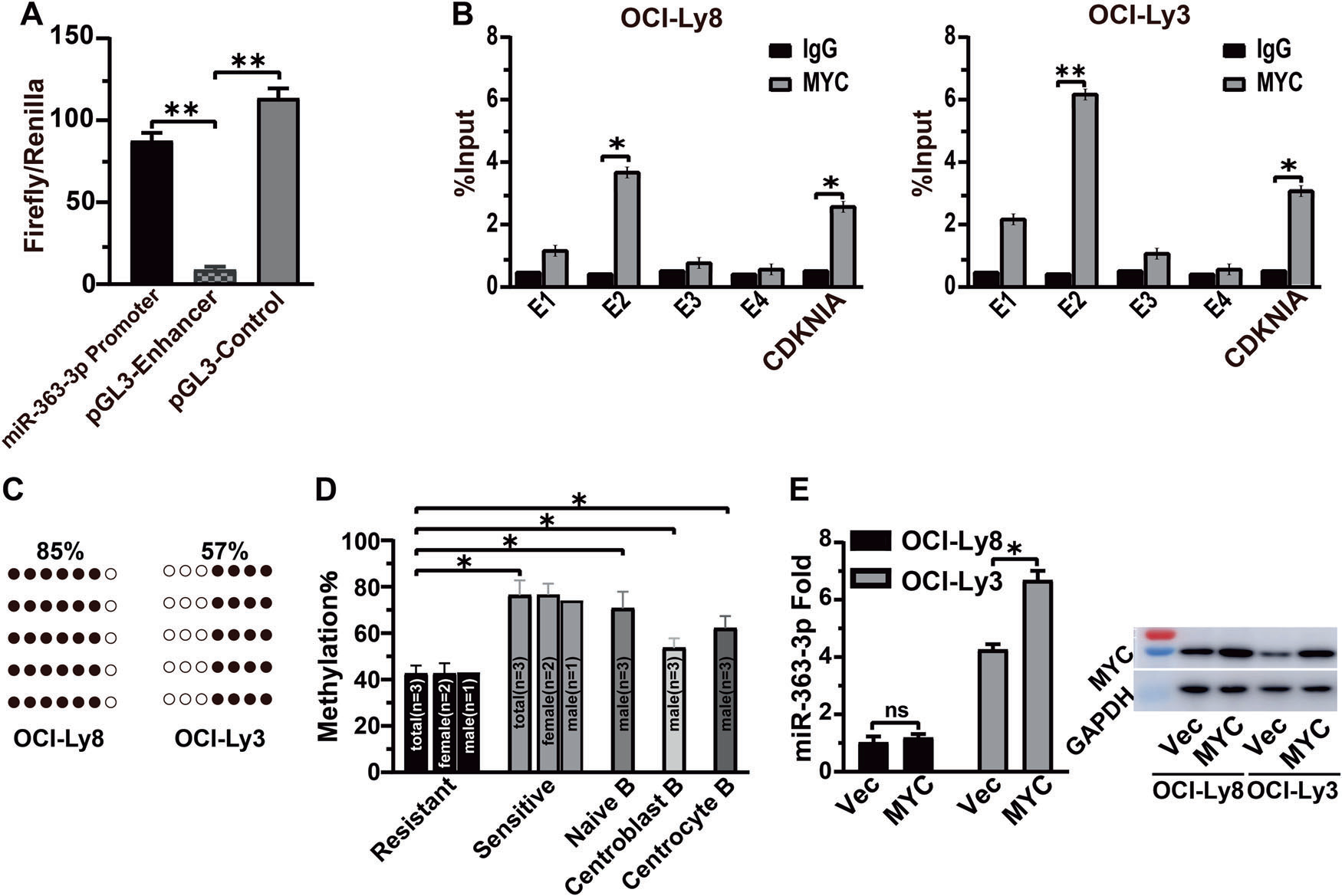
CpG岛周围1.258 kb的序列被预测为候选miRNA-363-3p启动子,其功能通过荧光素酶活性来验证(图2A)。生物信息学分析显示,在CpG岛上有四个MYC结合的E盒元件。通过染色质免疫沉淀+ PCR法确定E-box-2为MYC的精确结合位点。值得注意的是,在OCI-Ly3中MYC的结合水平高于OCI-Ly8(图2B)。亚硫酸氢盐测序PCR位点特异性DNA甲基化分析发现,OCI-Ly3中E-box-2周围的CpG甲基化低于OCI-Ly8 (图2C)。通过观察到耐药患者的甲基化水平低于敏感患者和naive、中心母细胞、中心细胞B细胞(图2D),进一步验证了甲基化对miRNA-363-3p表达的影响(图2D)。在耐药和敏感患者中,女性与男性的比例均为2:1,平衡了性别对x染色体DNA甲基化的影响,而且MYC的异位表达并没有显著增加OCI-Ly8中miRNA-363-3p的水平(图2E)。综上所述,这些结果证实了miRNA-363-3p除了MYC外,还受DNA甲基化的转录调控。
3)MiRNA-363-3p直接抑制DUSP10表达,然后增强JNK磷酸化
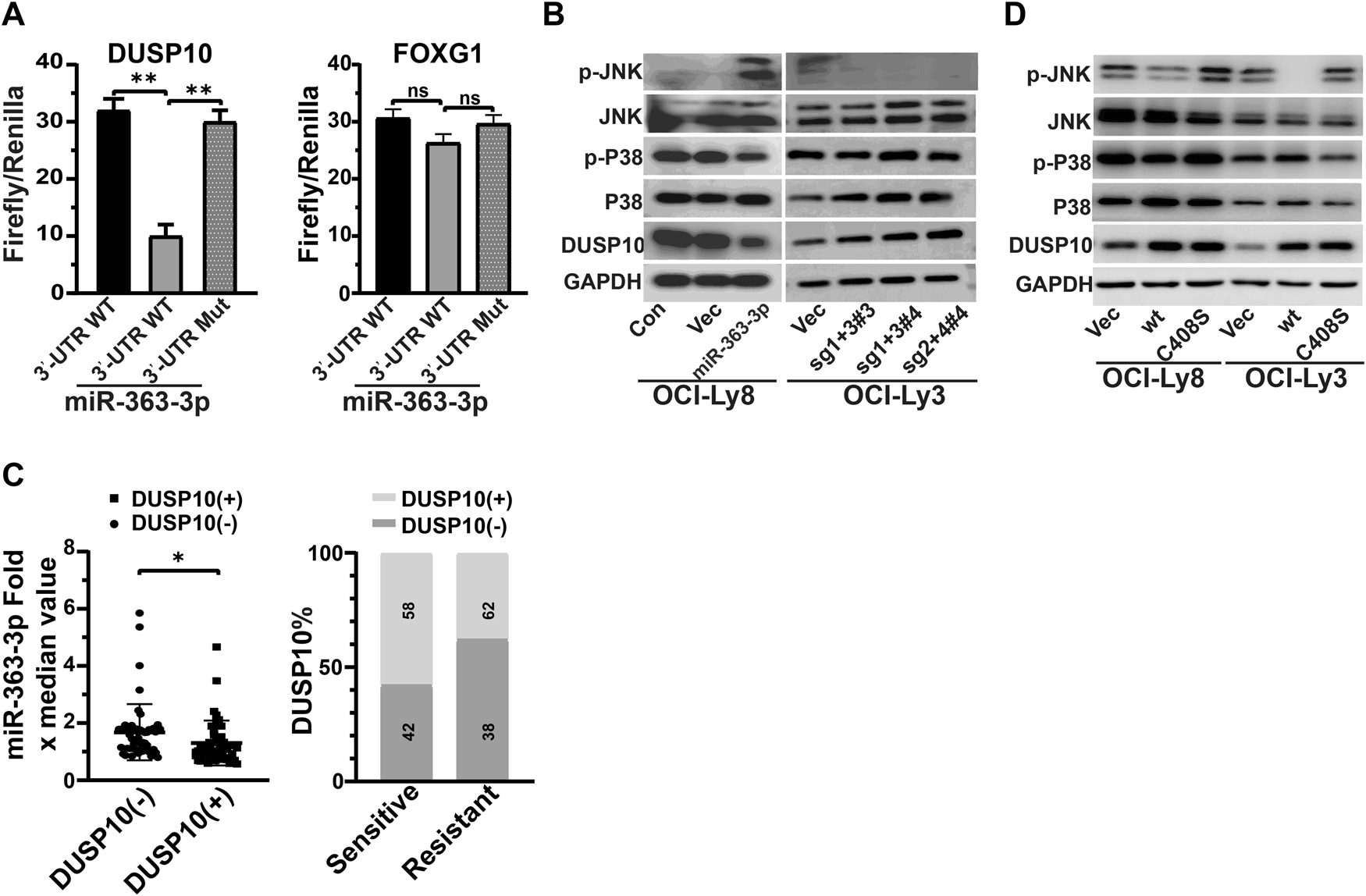
我们通过DLBCL患者的基因表达谱数据和软件预测确定了DUSP10、FOXG1和CACNA1C为候选靶基因。为了检测miRNA-363-3p对这些基因的直接调控,我们进行荧光素酶报告基因实验。结果表明,miRNA-363-3p模拟物显著抑制含有DUSP10和CACNA1C基因3′UTR的质粒的荧光素酶活性,但不抑制FOXG1基因的3′UTR(图3A)。据报道,CACNA1C会影响利妥昔单抗耐药性。在此,我们重点研究了DUSP10在化疗耐药性中的作用。Western blot分析证实,DUSP10蛋白在miRNA-363-3p-异位表达的OCI-Ly8中减少,而在miRNA-363-3p-敲除的OCI-ly3中增加(图3B)。106例DLBCL患者的DUSP10表达与miRNA-363-3p和化疗耐药性呈负相关(图3C)。据报道,JNK和p38受到DUSP10介导的MAPK信号通路的去磷酸化的负调控。我们发现,在miRNA-363-3p-异位表达的OCI-Ly8中,磷酸化JNK的水平增强,而在miRNA-363-3p-敲除OCI-Ly3中,磷酸化JNK的水平降低(图3B)。磷酸酶死亡的DUSP10 (C408S)进一步验证了miRNA-363-3p/DUSP10/JNK轴在DLBCL细胞中的功能(图3D)。
4)MiRNA-363-3p相关的耐药性与阿霉素诱导的DNA双链断裂(DSB)修复增强有关
我们对78个抗性相关上调基因进行David通路分析,发现DNA损伤修复途径是富集的,包括HR的负调节(RECQL5,POLQ)和DNA修复(FAAP100,RECQL5,DDX11,TICRR,PSME4,POLQ,SMC1A)(图4A)。γH2AX是已知的DSB生物标记物。miRNA-363-3p敲除导致OCI-Ly3细胞中阿霉素诱导的γH2AX水平增加(图4B)。此外,miRNA-363-3p表达的变化不影响TP53和BCL2蛋白的水平。正如预期的那样,ATF2和磷酸化ATF2的水平与miRNA-363-3p表达呈正相关(图4C)。DSB主要由HR和NHEJ修复,偶尔也由容易出错的替代NHEJ修复。使用报告质粒系统,我们证实miRNA-363-3p模拟物显著降低293 T细胞的HR活性,但增加NHEJ和选择性NHEJ活性;miRNA-363-3p抑制剂显示出相反的作用(图4D)。RAD51是HR修复的核心分子。免疫荧光分析发现,阿霉素诱导的RAD51病灶通过miRNA-363-3p的异位表达而明显减少,但通过miRNA-363-3p的敲除而增加(图4E)。

5)通过抑制JNK和PARP1逆转miRNA-363-3p相关的化疗耐药性
PARP1抑制剂已被证明对缺乏HR的癌细胞有显著疗效。因此,我们评估了pan-JNK抑制剂SP600125和PARP1抑制剂BGB-290克服DLBCL细胞中与miRNA-363-3p/DUSP10/JNK轴相关的化疗耐药性的潜力。结果表明,它们明显增强了高miRNA-363-3p表达的OCI-Ly3细胞中阿霉素诱导的γH2AX水平(图5A)。此外,在高表达miRNA-363-3p的细胞系中观察到阿霉素和SP600125或BGB-290之间的明显协同作用(图5B)。在携带OCI-Ly3细胞的异种移植小鼠模型中进一步证实了其疗效。添加SP600125或BGB-290可显著增加阿霉素诱导的肿瘤消退和存活率(图5C)。总之,我们证明,通过抑制DLBCL细胞中的JNK和PARP1,可以逆转miRNA363-3p相关的化疗耐药性。内在机制如图5D所示。
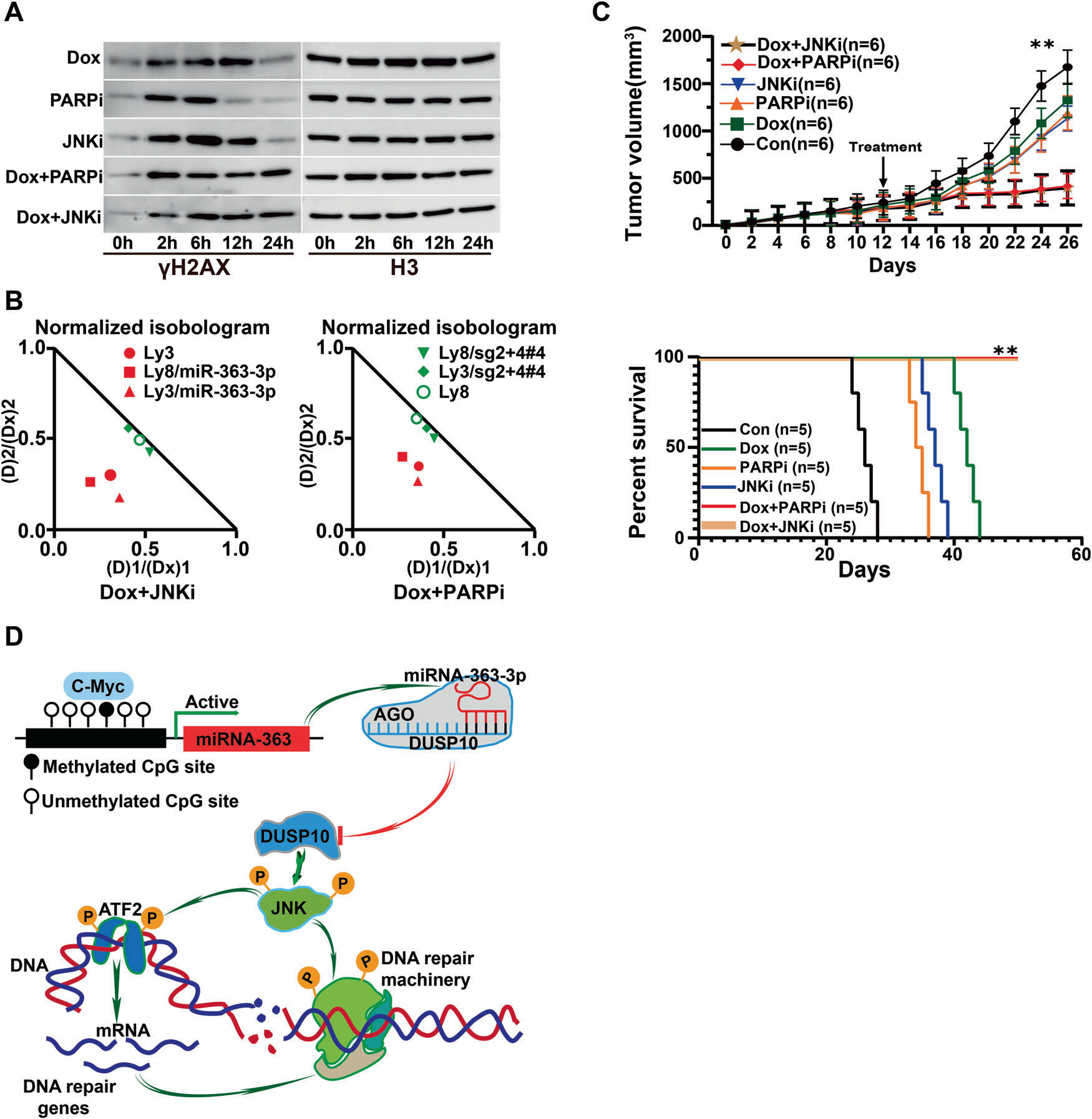
结论:我们创造性地提出了一种新的蒽环类耐药机制,涉及miRNA-363-3p/DUSP10/JNK介导的DSB修复增强。针对这种耐药机制的新方法为克服化学耐药性提供了潜在的选择。
参考文献:
Zhou W, Xu Y, Zhang J, Zhang P, Yao Z, Yan Z, Wang H, Chu J, Yao S, Zhao S, Yang S, Guo Y, Miao J, Liu K, Chan WC, Xia Q, Liu Y. MiRNA-363-3p/DUSP10/JNK axis mediates chemoresistance by enhancing DNA damage repair in diffuse large B-cell lymphoma. Leukemia. 2022 Apr 29. doi: 10.1038/s41375-022-01565-6.