






线粒体融合/裂变干预——特异性心脏病药物作者发现琥珀酸脱氢酶组装因子4(SDHAF4)在心肌和人类患者的病变心脏中在病理应激反应中下调,并证实心脏SDHAF4介导
琥珀酸脱氢酶被称为线粒体复合体II,因其在线粒体功能中的独特作用而吸引了人们对其参与人类疾病研究的兴趣。在此,作者发现琥珀酸脱氢酶组装因子4(SDHAF4)在心肌和人类患者的病变心脏中在病理应激反应中下调,并证实心脏SDHAF4介导的线粒体复合体II组装中断能促进扩张型心肌病进展。本研究于2022年6月发表杂《NATURE COMMUNICATIONS》IF:17.694。
技术路线:

主要实验结果:
1、人中SDHAF4异常表达以响应心脏损伤和心脏病变
心脏是线粒体含量较高的器官之一,心肌的功能主要依赖于线粒体的稳态。作者猜想SDHAFs可能参与了调控心脏功能。检测了心肌梗死(MI)小鼠模型心脏组织中的4个SDH组装因子的表达,结果发现只有SDHAF4的mRNA和蛋白表达(Fig. 1a-1d)在MI建模后显著下调。免疫荧光揭示SDHAF4与线粒体的初步共定位(Fig. 1e)。在酵母中SDHAF4可以促进SDHA与SDHB的组装。值得注意的是,MI手术后SDHA和SDHB之间的相互作用降低,提示SDHAF4在MI小鼠中起一定的下调作用(Fig. 1f, g)。在GSE135055数据中,SDHAF4在心脏中的表达下降与人类心脏疾病密切相关,包括DCM(扩张型心肌病)和MCD(微血管冠脉疾病)。综上所述,这些结果表明SDHAF4表达对心脏损伤非常敏感。作者推测SDHAF4可能是参与调节心脏稳态的复合物II的独特因子。

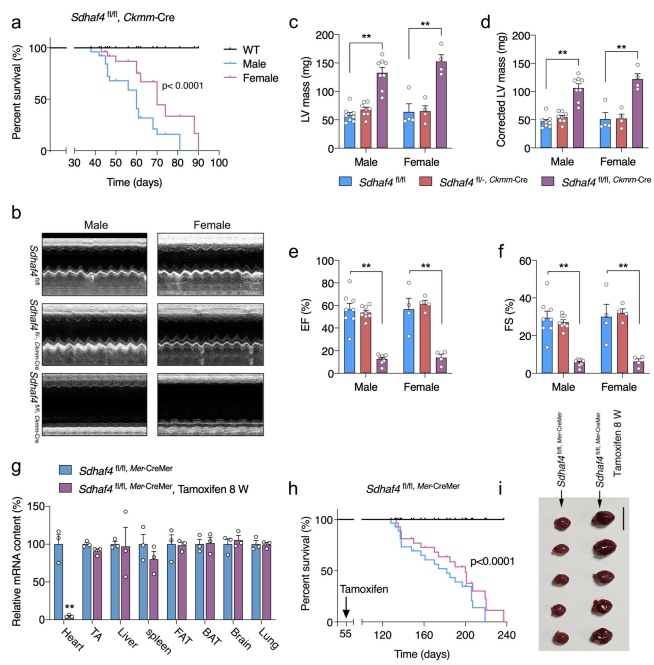
通过构建 Sdhaf4 的 floxed 等位基因(Sdhaf4fl)并随后生成 Ckmm-Cre 转基因介导心脏和骨骼肌中 Sdhaf4 失活的小鼠,以检测SDHAF4 在心脏中的作用。该突变小鼠命名为Sdhaf4 fl/fl,Ckmm-Cre或Sdhaf4 Ckmm。Sdhaf4 Ckmm小鼠的存活力显著降低,没有一只突变鼠存活超过12周(Fig. 2a)。Sdhaf4 Ckmm小鼠的心脏明显增大,提示突变鼠心脏缺陷。心电图结果显示和对照组比较,Sdhaf4 Ckmm小鼠的FS%和EF%都显著下降(Fig. 2c–f),表明Sdhaf4丢失损害心脏功能。
随后将Sdhaf4-floxed 等位基因小鼠和三苯氧胺诱导的Myh6-Cre转基因小鼠(Mer-CreMer)杂交以进一步验证SDHAF4缺失对心脏表型的影响。三苯氧胺处理完善了Cre重组酶介导的小鼠心肌组织中SDHAF4的特异性缺失(Fig. 2g)。三苯氧胺诱导8周后,纯合小鼠(Sdhaf4 fl/fl,Mer-CreMer,Sdhaf4 Mer-CreMer)的存活力显著下降,且心脏显著增大(Fig. 2h, i)。综上,这些结果表明SDHAF4缺失导致心脏和致命性异常,这表明Sdhaf4是心脏的一个必需基因。
3、心脏敲除SDHAF4后诱发扩张型心肌病
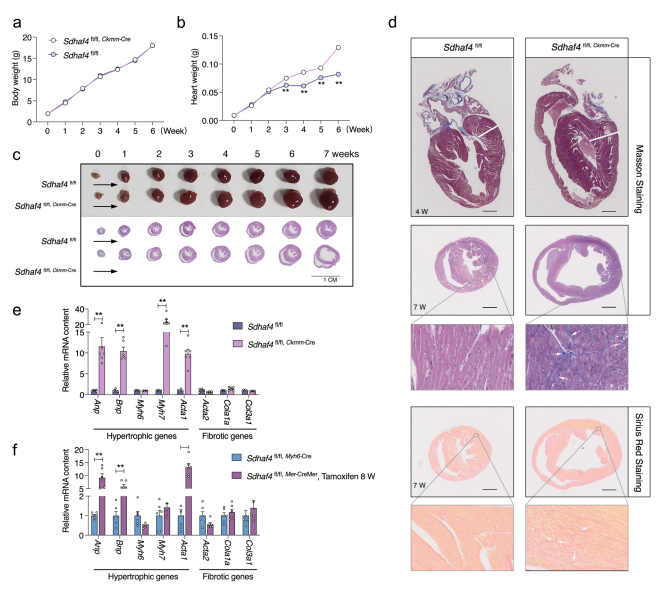
接下来作者分析了心脏在不同阶段的总体形态学以监测Sdhaf4 Ckmm小鼠的病理过程。在出生后的前6周,Sdhaf4 Ckmm小鼠的体重和它们同窝出生的WT小鼠的体重相似(Fig. 3a),但是心脏重量在3周的时候逐渐增加(Fig. 3b)。HE染色观察到最后阶段的心室组织发生扩张,表明Sdhaf4缺失导致心脏重塑和DCM进展(Fig. 3c)。Masson染色显示在最初4周内,Sdhaf4 Ckmm小鼠的心脏纤维化无明显改变,但是在后后7周出现明显的胶原沉积(Fig. 3d)。一致的,心肌病标记基因(胎儿基因)的表达在Sdhaf4 Ckmm小鼠4周龄时开始上调(Fig. 3e)。心肌病标记基因表达上调在三苯氧胺诱导的Sdhaf4 Mer-CreMer小鼠中也观察到了(Fig. 3f)。这些结果表明,心脏中Sdhaf4的缺失引发了心脏发病机制。
4、阻断复合体Ⅱ的组装导致线粒体功能异常
SDHAF4被证明与催化SDHA亚基特异性相互作用,并促进其与SDHB的结合,进而组装SDH复合物。所以作者检测了SDH复合物在小鼠心脏中不同时期的表达水平。SDHAF4的表达和其它SDH复合物亚基的表达在产后发育中逐渐增加(Fig. 4a)。在出生后3周龄时SDHAF4缺失导致SDH亚基的蛋白表达显著下降,但复合物I、III、IV和V组分的丰度在这个年龄的突变心脏中没有改变(Fig. 4b, c)。尽管Sdhaf4 Ckmm小鼠心脏中SDH亚基的蛋白表达下降,但是它们的mRNA水平并未改变(Fig. 4d),所以作者推测,复合体II的异常很可能发生在翻译后水平,主要是由于装配缺陷。
接下来作者分析了SDHAF4缺失对复合物组装的影响。使用心肌组织的免疫沉淀显示Sdhaf4缺失减少了SDHA和SDHB之间的相互作用(Fig. 4e, f),这个可能导致SDH亚基降解,泛素化修饰水平增加证实了这一猜想(Fig. 4g)。此外,线粒体超复合物的形成被抑制,并在突变心脏中观察到线粒体耗氧量的显著下降(Fig. 4h–j)。有趣的是,出生3周后,相对ATP水平保持不变,但在8周龄的Sdhaf4 Ckmm小鼠心脏中下降(Fig. 4k)。这些结果说明SDHAF4缺失阻断复合体Ⅱ的组装并导致线粒体呼吸异常。
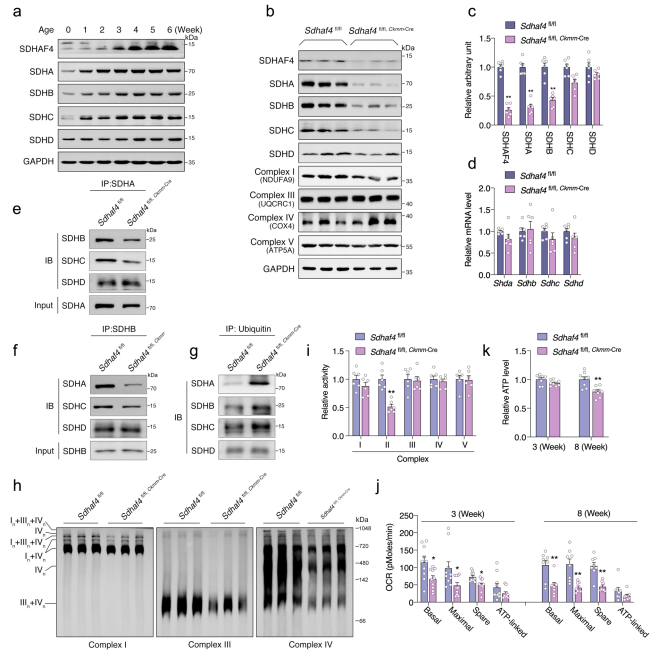
图4敲除SDHAF4后阻断复合体Ⅱ的组装并促进线粒体功能异常
鉴于复合体II是耦合线粒体电子传递链和TCA循环的关键因素,所以作者接下来探究SDHAF4缺失是否会导致TCA循环相关关键酶和代谢物的丰度。突变体早期(出生后3周)心脏中TCA循环相关关键酶的蛋白质水平与对照组没有区别,而在8周后,心脏中这些酶的水平整体下降(Fig. 5a, b)。此外,TCA循环相关代谢物水平在突变体小鼠中显著下降,包括丙酮酸,柠檬酸,异柠檬酸,苹果酸,富马酸,只有琥珀酸的丰度增加了(Fig. 5c)。这些变化是SDH活性丧失所导致的主要影响,而SDH活性是催化琥珀酸氧化为富马酸盐所必需的,这是TCA循环的关键步骤。进一步的代谢组学分析显示,SDHAF4缺失影响多个氨基酸代谢通路,其中精氨酸(Arg)生物合成通路改变最显著(Fig. 5d, e)。TCA循环和精氨酸生物合成之间的关联近来已有报道。精氨酸通过瓜氨酸-一氧化氮途径再生,瓜氨酸和天门冬氨酸(Asp)通过精氨酸琥珀酸合成酶(ASS)和精氨酸琥珀酸裂解酶(ASL)转化为精氨酸,富马酸作为副产物,而富马酸酶作用于富马酸的产物苹果酸转化为草酰乙酸生成天门冬氨酸,加入精氨酸的生物合成循环(Fig. 5f)。因此,SDHAF4的缺失不仅主要改变了TCA循环相关代谢物的水平,还抑制了精氨酸的再生,进一步加剧了代谢应激。

图5敲除SDHAF4后加速TCA循环相关代谢能力
5、阻断复合体II的组装改变心脏线粒体融合/裂变动力学
尽管3周龄Sdhaf4 Ckmm小鼠心脏的线粒体功能受损,但是细胞器的形态并未发生改变(Fig. 6a)。在TEM电镜中,Sdhaf4 Ckmm小鼠和对照组线粒体数目及其亚细胞分布无差异(Fig. 6b–e)。然而,随着病理性进展加剧,8周龄Sdhaf4 Ckmm小鼠心脏线粒体表现出明显的形态异常,包括肌原纤维/肌节组织异常,线粒体内部结构异常,如嵴畸形,电子密集的包裹体和低密度的隔室(Fig. 6f, g);此外,线粒体总数显著增加但线粒体大小显著减小(Fig. 6h–j)。
接下来探究Sdhaf4 Ckmm小鼠心脏中线粒体数量增加但大小减小是否是由于线粒体生物发生和融合过程改变导致的。然而,qPCR分析显示Sdhaf4 Ckmm小鼠心肌中mtDNA的相对丰度降低,而不是增加(Fig. 6k)。PGC-1α和其它线粒体生物发生调节基因的表达在Sdhaf4 Ckmm小鼠中也都下调表达(Fig. 6l, m)。因此,这些细胞器数量的增加不太可能是由于线粒体生物发生的改变造成的。所以接下来作者测定了调节线粒体融合和裂变的多种因子的蛋白质水平,但未检测到对照组和突变心脏之间的显著差异。但是,Drp1的转录后修饰在Sdhaf4 Ckmm小鼠中显著改变,该基因是线粒体融合的关键基因。Drp1的激活高度依赖于两个丝氨酸位点:丝氨酸616位点磷酸化(p-Drp1 s616)增强线粒体融合,丝氨酸637位点磷酸化(p-Drp1 s637)抑制线粒体融合活性。作者发现在3周大的Sdhaf4 Ckmm小鼠中p-Drp1 s616增加而p-Drp1 s637下降(Fig. 6n)。Drp1的激活在8周大的Sdhaf4 Ckmm小鼠中也观察到了(Fig. 6n, p),提示在SDHAF4缺失的情况下,Drp1在早期且持续激活。综上所述,这些结果表明SDHAF4缺乏可能会增强线粒体裂变,导致细胞器碎裂,因而可能是线粒体数量增加的原因。
线粒体自噬通常与线粒体融合/裂变动力学耦合,以使受损细胞器回收和清除。有趣的是,自噬激活的标志LC3-II的水平在8周龄的Sdhaf4 Ckmm小鼠心脏中显著增加(Fig. 6o, p)。使用荧光报告基因(mt-Keima)方法评估H9C2大鼠心肌细胞线粒体自噬水平,证实了SDHAF4缺失导致线粒体自噬小体形成增加(Fig. 6q)。这些表明SDHAF4缺失导致了心肌细胞的线粒体自噬。
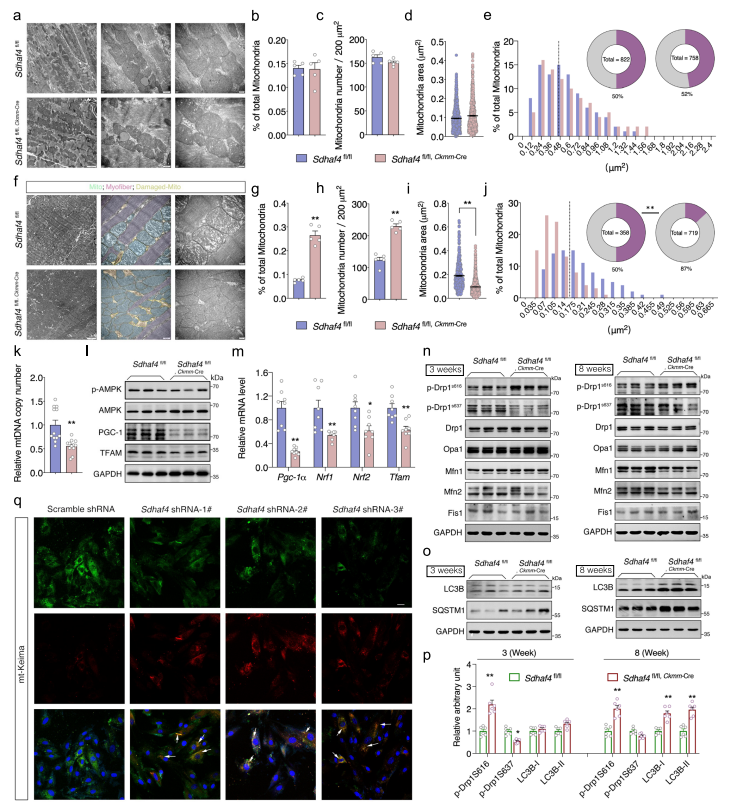
图6阻断复合体Ⅱ的组装促进心脏线粒体自噬
6、线粒体靶向干预延长DCM小鼠的寿命
基于前面的观察,作者猜想代谢能力受损和线粒体自噬加速可能是DCM的Sdhaf4Ckmm小鼠中的两个主要线粒体事件,因此,靶向代谢或药物干预可能会有所疗效。为验证该猜想,作者使用口服水富马酸钠或腹腔注射特异性Drp1抑制剂Mdivi-1处理小鼠。线粒体和肌纤维完整性通过补充富马酸盐或Mdivi-1得到改善(Fig. 7a)。线粒体数量和平均大小也基本恢复(Fig. 7b, c)。心脏肥厚和纤维相关基因,Anp的mRNA表达在两个干预组中都显著下降,Bnp和Myh7的上调也被Mdivi-1处理抑制(Fig. 7d)。心电图显示Mdivi-1处理显著改善Sdhaf4Ckmm小鼠的心脏功能,表现为EF%和FS%上升(Fig. 7e–j)。重要的是,补充富马酸盐或Mdivi-1处理都显著延长了小鼠的存活时间(Fig. 7k)。表明以TCA代谢或线粒体动力学为靶点的方法是治疗心功能不全的有效方法。
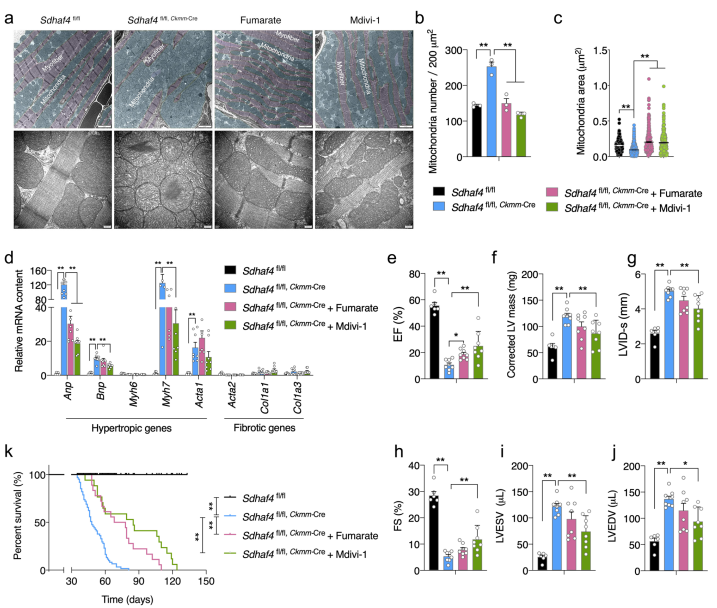
图7抑制Drp1或补充富马酸可延长扩张型心肌病小鼠的寿命
由于富马酸是SDH催化的直接产物,因此富马酸丢失可能是Sdhaf4 Ckmm小鼠心脏的关键特征。值得注意的是富马酸的下调和琥珀酸的上调都在MI小鼠中报道过,表明富马酸盐相关代谢异常可能是心脏发病的关键过程之一。为了验证该猜想,对MI小鼠术前术后补充富马酸,结果显示,与MI小鼠比较,富马酸补充的MI小鼠的心脏重量/体重比显著下降,EF%和FS%显著增加,心脏肥厚和纤维相关基因也显著回复(图8)。这些结果说明直接补充富马酸可改善MI小鼠的心脏功能。综上所述,这些观察结果提示MI患者SDHAF4相关富马酸缺失在心功能障碍的进展中起着至关重要的作用。
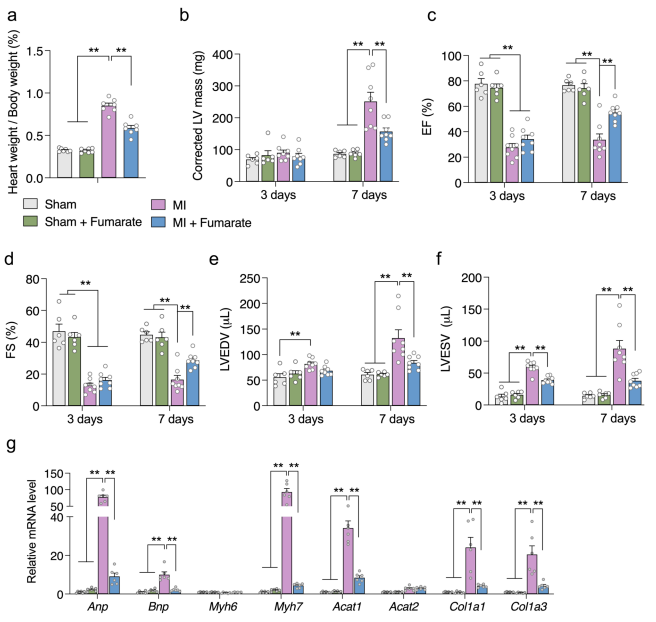
图8 补充富马酸可以改善MI小鼠的心功能
综上所述,本文研究了SDHAF4在小鼠体内的病理参与,以确定其在维持心功能中的重要作用。SDHAF4是新近发现的复杂II组装因子,其表达对应激敏感。SDHAF4的缺失破坏了复合物II的组装,促进了SDH单位的降解,从而逐步导致代谢障碍和线粒体自噬过量,最终导致DCM和心力衰竭。此外,针对线粒体代谢和动力学的干预是改善心脏功能的有效策略。小鼠SDHAF4蛋白的生理评价为复杂II缺陷相关DCM的发展和临床实践提供了更多的见解。
参考文献:
Wang Xueqiang., Zhang Xing., Cao Ke., Zeng Mengqi., Fu Xuyang., Zheng Adi., Zhang Feng., Gao Feng., Zou Xuan., Li Hao., Li Min., Lv Weiqiang., Xu Jie., Long Jiangang., Zang Weijin., Chen Jinghai., Ding Jian., Liu Jiankang., Feng Zhihui.(2022). Cardiac disruption of SDHAF4-mediated mitochondrial complex II assembly promotes dilated cardiomyopathy. Nat Commun, 13(1), 3947. doi:10.1038/s41467-022-31548-1