






抑制HDAC3通过增强CXCL10介导的趋化性和免疫细胞募集促进抗肿瘤免疫
实验方法:细胞增殖和活力分析,组织分离,免疫荧光染色,ELISA,ELISpot (酶联免疫斑点测定法),肿瘤浸润性CD4+和CD8+ T细胞的大量RNA测序及分析,肿瘤RNA测序及分析,qRT-PCR,ChIP-seq和ChIP-qPCR检测,流式细胞术,Western blot,TMA的IHC测量与分析
组蛋白去乙酰化酶(histone deacetylase, HDAC)抑制剂是一类新的抗癌药物,一般认为其抗肿瘤活性是通过直接引起肿瘤细胞周期阻滞和细胞凋亡来发挥的。然而,我们证明了I类HDAC抑制剂,如Entinostat和Panobinostat,可以有效抑制免疫正常而非免疫缺陷小鼠的肿瘤生长。对Hdac1, 2, 3敲除肿瘤细胞的进一步研究表明,HDAC3的肿瘤特异性失活通过激活抗肿瘤免疫来抑制肿瘤生长。具体来说,我们发现HDAC3可以直接结合到启动子区域,抑制CXCL9, 10, 11化学因子的表达。Hdac3缺陷的肿瘤细胞高水平表达这些趋化因子,通过将CXCR3+ T细胞募集到肿瘤微环境(TME)中,抑制免疫能力小鼠的肿瘤生长。此外,HDAC3与CXCL10在肝细胞癌肿瘤组织中的表达呈负相关,也提示HDAC3可能参与抗肿瘤免疫调节和患者生存。因此,抑制HDAC3通过增强免疫细胞向TME的浸润来抑制肿瘤生长。这一抗肿瘤机制可能有助于指导基于HDAC3抑制剂的治疗。
技术路线:

结果:
(1) HDAC抑制剂以免疫依赖的方式抑制肿瘤生长
HDAC抑制剂已被广泛用于癌症治疗。其中,Panobinostat(也称为LBH589,一种泛HDAC抑制剂)和Entinostat(也称为MS275, HDAC1和3选择性抑制剂)正在进行III期临床试验。我们首先检测了恩替诺他和帕比诺他对MC38小鼠结肠腺癌和MCA205纤维肉瘤肿瘤生长的影响。我们发现Entinostat和Panobinostat在免疫正常的C57BL/6小鼠中有效抑制MC38和MCA205肿瘤的生长(图1A-D),但在免疫缺陷的裸小鼠中无效(图1E-H)。这些结果表明,I类HDAC抑制剂Entinostat和Panobinostat以免疫依赖的方式抑制MCA205和MC38肿瘤的生长。
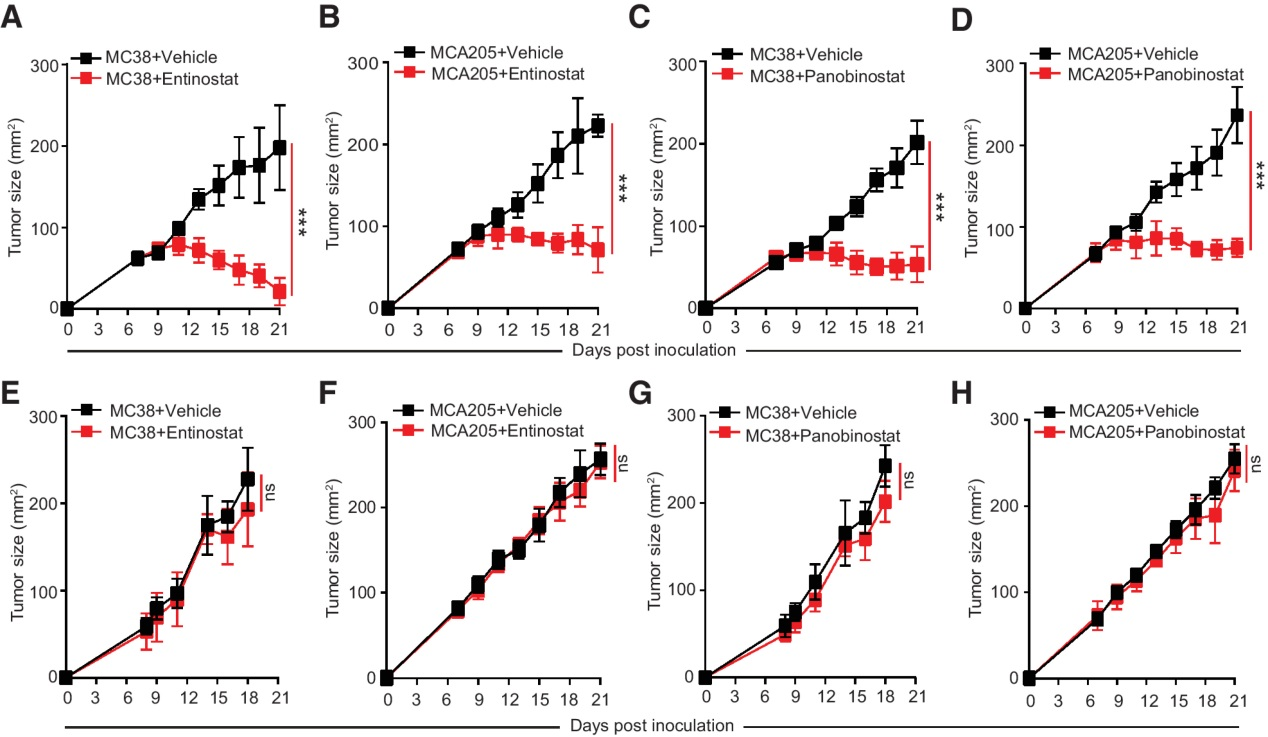
图1:HDAC抑制剂以免疫依赖的方式抑制肿瘤生长
(2) Hdac3缺乏抑制肿瘤生长,而肿瘤生长依赖于宿主免疫反应
Panobinostat和Entinostat均抑制MCA205和MC38肿瘤的生长,提示抑制一种或多种I类HDAC可能抑制肿瘤生长。为了探索Hdac1, 2, 3基因在肿瘤生长中的作用,我们利用CRISPR/Cas9技术敲除MC38细胞中的Hdac1, 2, 3基因(补充图S1A-S1C),并将细胞注射到C57BL/6小鼠体内。比较Hdac1−/−,Hdac2−/−和Hdac3−/−我们发现Hdac3缺失显著抑制MC38肿瘤的生长,而Hdac1和Hdac2缺失对MC38肿瘤的生长无明显影响(图2A)。为了证实Hdac3基因在肿瘤生长中的作用,我们进一步敲除(Hdac3+/−)和敲除(Hdac3−/−) MC38和MCA205细胞中的Hdac3基因。比较Hdac3+/−和Hdac3−/−我们发现,在C57BL/6小鼠和裸鼠中,敲低和敲除MC38和MCA205肿瘤及其相应的WT肿瘤均显著抑制了C57BL/6小鼠中MC38和MCA205肿瘤的生长(图2B和C),而在裸鼠中则没有抑制作用(图2D和E)。这些结果表明,HDAC3促进肿瘤生长并依赖于宿主免疫应答。
将MCA205细胞静脉注射到小鼠体内后,肿瘤会发展并转移到多个器官,导致小鼠死亡。然后我们比较静脉注射WT或Hdac3−/−的C57BL/6小鼠的存活率。MCA205细胞。移植WT MCA205细胞的小鼠在移植后不能存活超过17天,而所有移植Hdac3−/− MCA205细胞存活(图2F),表明Hdac3缺失减少了转移并保护小鼠免于转移相关死亡。我们还分析了GEO和TCGA数据库中876例胃癌患者Hdac1, 2, 3基因表达与OS的相关性。Hdac3基因的高表达与胃患者较低的OS相关,而Hdac1和Hdac2基因的高表达与胃患者较高的OS相关(图2G-I)。此外,HDAC3 mRNA在各种癌症组织中的表达均高于相应的正常组织,包括胆管癌、宫颈鳞状细胞癌和宫颈内膜腺癌、肺腺癌、肺鳞状细胞癌(补充图S2)。这些结果提示特异性抑制Hdac3可能通过抗肿瘤免疫抑制肿瘤生长。
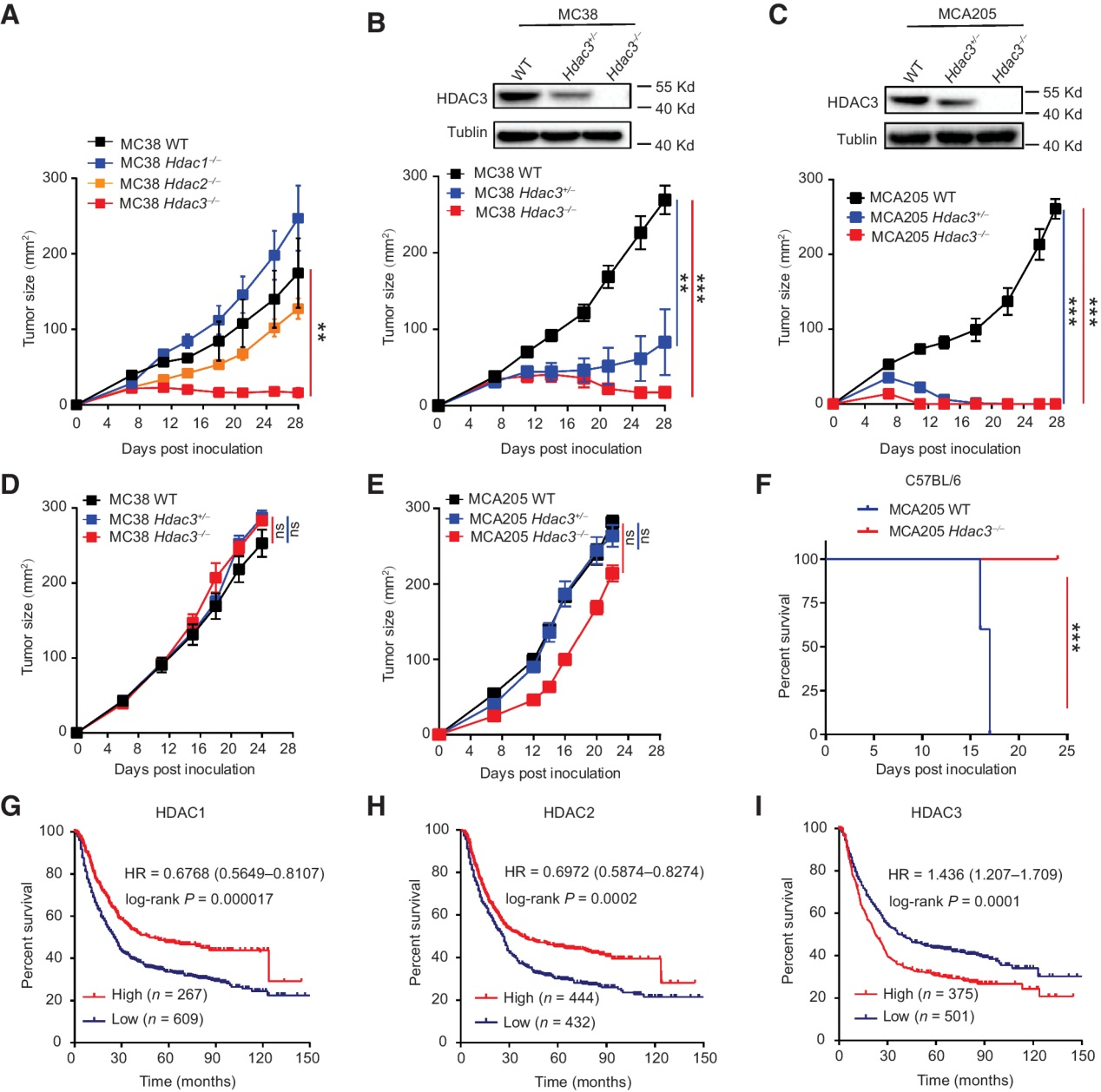
图2:Hdac3缺乏抑制肿瘤生长并依赖于宿主免疫反应
(3) Hdac3缺陷肿瘤的局部免疫反应增强
CCK-8 (补充图S3A和S3B)、Ki-67染色(补充图S3C和S3D)和单克隆形成(补充图S3E和S3F)检测显示,Hdac3缺乏对MC38和MCA205细胞的活力和增殖没有影响。为了探索HDAC3是否在调节局部TME中发挥作用,我们使用流式细胞术检测分析了WT和Hdac3+/− MCA205肿瘤中的肿瘤浸润淋巴细胞(TIL),因为Hdac3−/−细胞没有形成足够大的肿瘤进行流式细胞术分析。事实上,我们观察到肿瘤浸润的CD4+、CD8+、CD11c+、CD11b+F4/80+、CD4+ IFNγ+和CD8+ IFNγ+免疫细胞的数量增加。MCA205肿瘤(图3A;补充图S4)。结果表明,当肿瘤细胞特异性缺乏Hdac3时,更多抗原呈递DCs和肿瘤杀伤T细胞被招募到MCA205 TME中。为了更好地计数肿瘤浸润T细胞的大小和分布,对WT、Hdac3+/−和Hdac3−/− MCA205肿瘤进行免疫荧光染色。更多的CD4+、CD8+和IFNγ+细胞浸润Hdac3+/−和Hdac3−/− MCA205肿瘤与WT型MCA205肿瘤的比较(图3B, C;补充图S5A和S5B)。还采用ELISpot法评估WT和Hdac3+/− MCA205稳定表达的鸡卵细胞中的OVA特异性IFNγ分泌T细胞的频率。结果显示,Hdac3+/− MCA205肿瘤中IFNγ分泌T细胞比WT肿瘤中更多(图3D和E)。这些结果表明,Hdac3缺陷肿瘤中浸润T细胞的数量和特征明显不同于WT肿瘤。
为了表征TME中浸润性T细胞的特征,我们通过FACS从WT和Hdac3+/− MCA205肿瘤中分离CD45+CD4+ and CD45+CD8+ T细胞,并进行大量RNA测序检测基因表达。与WT细胞相比,从Hdac3+/−肿瘤分离的T细胞表达更高水平的转录因子,如Eomes和Tcf712;细胞表面受体如Sell、Vsir和Cd28;效应分子如Gzmb、Ifng、Prf1、Tnf;和耐受性相关基因如Nr4a2和Egr2 (图3F)。以上结果表明,肿瘤细胞特异性Hdac3缺失引发了免疫细胞的差异浸润,在TME中比在WT肿瘤中更强的局部免疫反应。
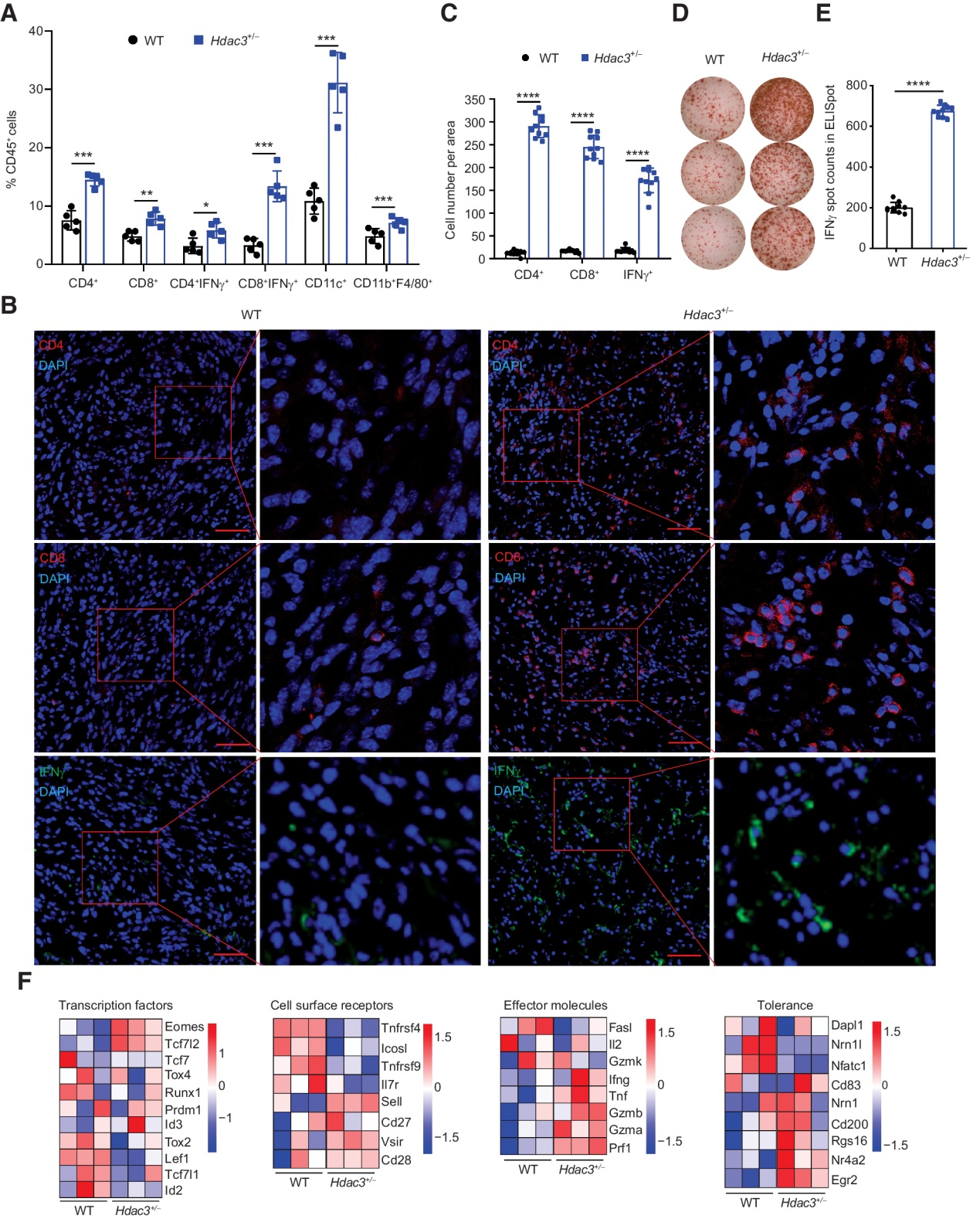
图3:Hdac3缺陷肿瘤的局部免疫反应增强
(4) Hdac3缺乏上调趋化因子信号通路和T细胞受体信号通路
大量RNA-seq结果(图3F)显示,HDAC3可能通过调节TME中免疫细胞(尤其是T细胞)的浸润和特征来影响肿瘤生长。为了探索HDAC3调节T细胞浸润和TME的机制,我们对WT和Hdac3+/− MCA205肿瘤组织进行RNA测序。KEGG注释分类分析比较Hdac3+/−和WT肿瘤显示,Hdac3缺失显著上调趋化因子信号通路、T细胞受体(TCR)信号通路和细胞因子-细胞因子受体相互作用过程中的基因(图4A和B)。GSEA还显示,趋化因子信号通路(图4C)、细胞因子-细胞因子受体相互作用(图4D)和TCR信号通路(图4E)在Hdac3缺失的MCA205肿瘤中显著富集。这些结果表明,与T细胞募集相关的TCR信号通路和趋化因子信号通路可能有助于抑制Hdac3缺陷MCA205细胞的肿瘤生长。然后,我们选择了与T细胞和T细胞募集相关的基因,并评估了它们在WT和Hdac3+/− MCA205肿瘤中的相对表达。丰富的T细胞标记物(Cd3d、Cd3e、Cd3g、Cd4、Cd8a、Ctla4、Zap70和Cxcr3)和T细胞募集相关基因(Cxcl9, 10, 11)在Hdac3缺陷肿瘤中显著上调(图4F)。为了验证RNA测序的结果,通过qRT-PCR将Hdac3−/− MCA205肿瘤与WT肿瘤中的基因表达进行比较。我们证实,Hdac3缺陷导致T细胞标记和T细胞招募相关基因上调(补充图S5C)。这些结果使我们假设HDAC3可能通过降低趋化因子如Cxcl9, 10, 11的表达来抑制T细胞募集到TME。
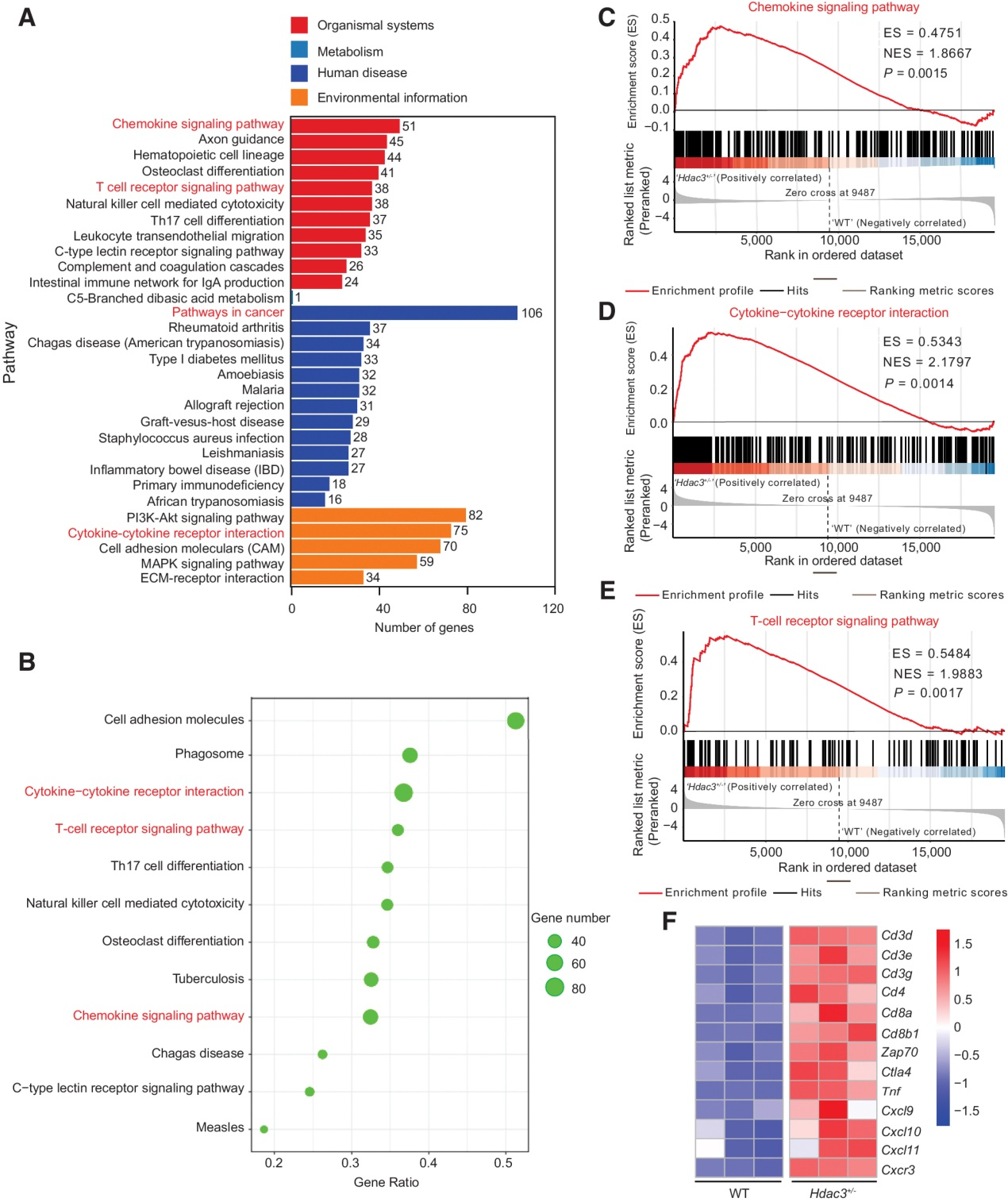
图4:Hdac3缺乏上调趋化因子信号通路和TCR信号通路
(5) HDAC3下调IFN诱导的趋化因子Cxcl9, 10, 11的表达
趋化因子Cxcl9, 10, 11是IFN诱导基因,IFN信号通路在抗肿瘤免疫应答中起重要作用。因此,我们定量分析了在IFNα-,IFNβ-和IFNγ处理的WT和Hdac3−/− MCA205细胞中Cxcl9, 10, 11的mRNA的表达。qRT-PCR结果显示,与WT型细胞相比,IFN处理的Hdac3−/−细胞中Cxcl9, 10, 11 mRNA表达水平明显升高(图5A)。接下来,我们通过ELISA检测在IFNα-,IFNβ-和IFNγ处理的WT和Hdac3−/− MCA205细胞培养上清中CXCL9, 10, 11的浓度。与mRNA表达相似,与WT细胞相比,在Hdac3−/− MCA205细胞培养的上清中CXCL9, 10, 11蛋白浓度也更高(图5B)。为了证实Cxcl9, 10, 11在体内Hdac3+/− MCA205肿瘤中表达上调,我们用ELISA法检测了肿瘤组织中CXCL9, 10, 11的浓度,并发现CXCL9, 10, 11在Hdac3+/− MCA205肿瘤的表达也高于WT MCA205肿瘤(图5C)。这些结果表明,Hdac3缺失上调了肿瘤细胞中Cxcl9, 10, 11的表达。
为了进一步证实HDAC3去乙酰化酶活性对Cxcl9, 10, 11表达的作用,我们在IFN处理的MCA205细胞中加入I类HDAC抑制剂Entinostat和HDAC3特异性抑制剂RGF966,通过qRT-PCR检测Cxcl9, 10, 11 mRNA的表达。HDAC抑制剂Entinostat和RGF966也诱导Cxcl9, 10, 11 mRNA表达上调(图5D)。相应地,我们也检测到Cxcl9, 10, 11受体基因Cxcr3在Hdac3+/− MCA205肿瘤中表达上调(图5E)。这些结果表明,HDAC3调节Cxcl9, 10, 11 mRNA的表达,并依赖于其去乙酰化酶的活性。
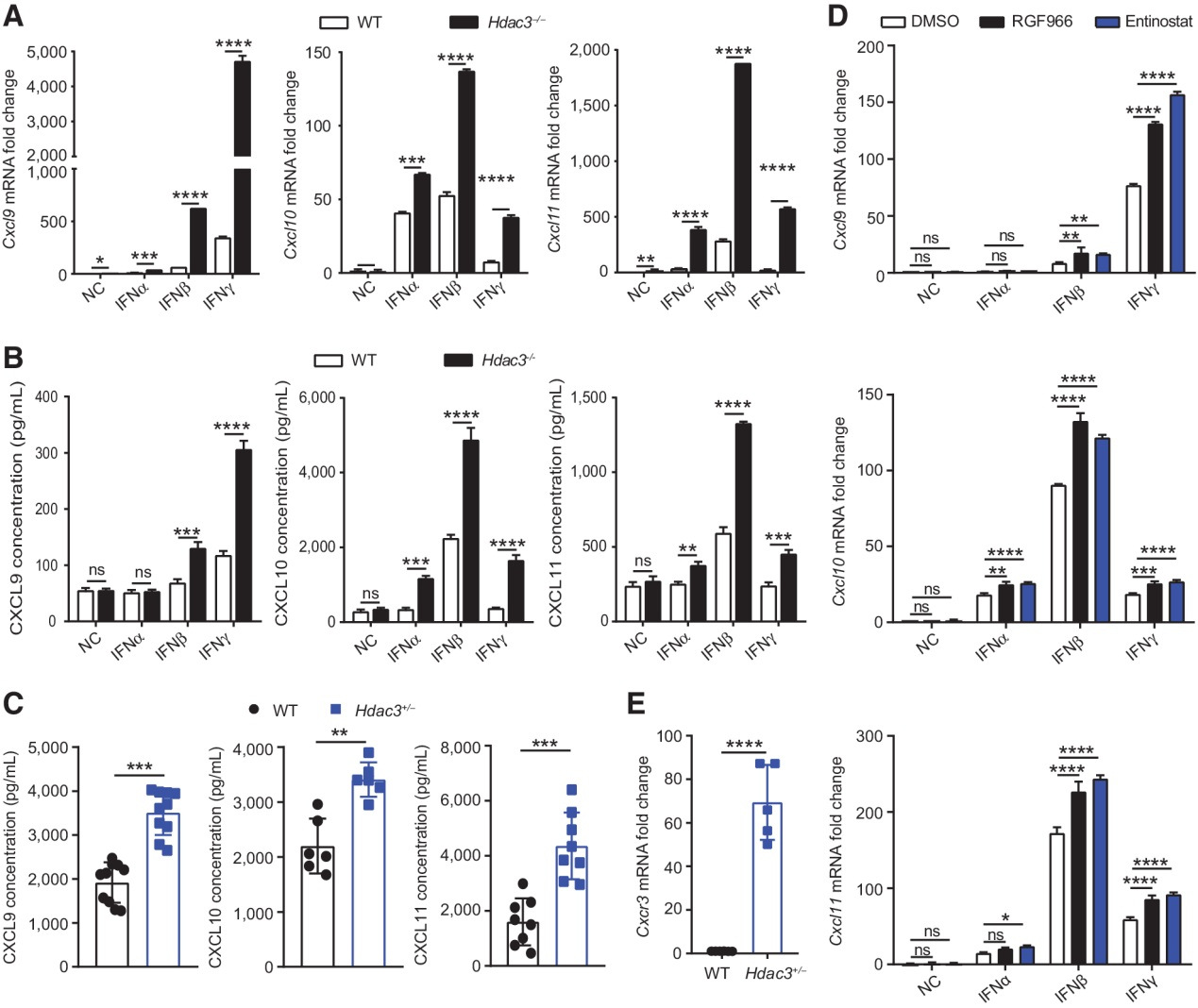
图5:HDAC3下调IFN诱导的趋化因子Cxcl9, 10, 11的表达
(6) HDAC3直接结合并使Cxcl10基因启动子去乙酰化
我们试图探索HDAC3调控Cxcl9, 10, 11表达的机制。首先,我们比较了IFNα-、IFNβ-和IFNγ处理与未处理的MCA205细胞中Cxcl9, 10, 11 mRNA的相对表达。在MCA205细胞中,Cxcl10的表达高于Cxcl9和Cxcl11 (补充图S6A和S6B),表明Cxcl10是MCA205肿瘤中负责CXCR3+ T细胞募集的主要趋化因子。HDAC3是一种表观遗传调节剂,可以介导组蛋白的去乙酰化修饰。接下来,我们通过ChIP-seq和ChIP-qPCR检测了HDAC3和H3K27乙酰化(H3K27ac)在Cxcl10启动子上的富集。在未处理和IFNβ刺激的WT MCA205细胞中,HDAC3在Cxcl10启动子区域富集,H3K27ac水平较低。然而HDAC3+/−和HDAC3−/− MCA205细胞中Cxcl10启动子上的HDAC3富集减少,IFNβ刺激后H3K27ac水平升高(图6A-D)。这些结果表明HDAC3可以结合并抑制Cxcl10启动子的H3K27ac。为了进一步确定HDAC3去乙酰化酶活性对Cxcl10转录的影响,我们用野生型HDAC3和去乙酰化失活突变体HDAC3−/− MCA205细胞(H134/135Q;补充图S7A和S7B)。CCK-8 (补充图S7C)、Ki67染色(补充图S7D)和单克隆形成实验(补充图S7E和S7F)显示,Hdac3过表达对MCA205细胞的活力和增殖没有影响。此外,ChIP-qPCR和ChIP-seq结果显示,WT HDAC3而不是HDAC3 H134/135Q突变体可以结合并抑制Cxcl10启动子的乙酰化(图6E-H)。进一步分析野生型和突变型HDAC3重组的HDAC3−/− MCA205细胞中Cxcl10 mRNA的表达,野生型HDAC3抑制Cxcl10 mRNA的表达,而H134/135Q-突变型HDAC3对Cxcl10 mRNA的表达无影响(图6I)。此外,我们随后将HDAC3−/− MCA205细胞接种到C57BL/6和裸鼠中,这些细胞由载体、野生型或H134/135Q突变型HDAC3重组。肿瘤生长监测显示,在H134/135Q突变体中,而不是在H134/135Q突变体中,HDAC3挽救了肿瘤的生长(图6J和K)。这些结果表明,HDAC3通过结合Cxcl10启动子和去乙酰化附近的组蛋白来抑制Cxcl10基因的表达。

图6:HDAC3直接结合并脱乙酰化Cxcl10基因启动子
(7) CXCR3抗体阻断以免疫依赖的方式拯救Hdac3缺陷的MCA205肿瘤生长
为了进一步验证Hdac3缺乏促进CXCR3+ T细胞募集来介导抗肿瘤免疫应答的假设,我们使用CXCR3抗体在WT和HDAC3+/− MCA205肿瘤中阻断CXCL9, 10, 11/CXCR3趋化因子信号通路。虽然TNFα抗体和IgG对HDAC3+/− MCA205肿瘤的缓慢生长无影响,但CXCR3抗体使HDAC3+/− MCA205肿瘤的生长恢复到与WT MCA205肿瘤相似的速度(图7A)。通过流式细胞术进一步分析WT,IgG治疗或CXCR3抗体治疗的HDAC3+/− MCA205肿瘤的TILs,结果显示,给予CXCR3抗体后,HDAC3+/− MCA205肿瘤中CD4+、CD8+、CXCR3+、CD4+CXCR3+和CD8+CXCR3+浸润免疫细胞减少(图7B)。免疫荧光染色结果显示,与IgG组相比,CXCR3抗体组HDAC3+/− MCA205肿瘤中CD4+、CD8+和IFNγ+细胞数量减少(图7C和D)。在Cxcr3抗体处理的HDAC3+/− MCA205肿瘤中,Cxcr3 mRNA也降低,这可能是由于表达Cxcr3的T细胞浸润减少(图7E)。这些结果表明,Hdac3缺陷肿瘤的生长降低可能是由于CXCR3+ T细胞浸润到TME介导的抗肿瘤活性增强。总之,我们的研究表明,在TME中,HDAC3通过调节Cxcl9, 10, 11基因的表达和Cxcl9, 10, 11-CXCR3轴介导的T细胞募集参与抗肿瘤免疫,为HDAC3的抑制提供了重要的抗肿瘤机制(图7F)。
我们还将MTX处理的WT、HDAC3+/−或HDAC3−/− MCA205细胞接种到左侧携带WT MCA205肿瘤的小鼠右侧,持续7天。我们发现,一侧MTX处理的HDAC3+/−和HDAC3−/− MCA205细胞连接抑制了另一侧WT MCA205肿瘤的肿瘤生长(补充图S8A和S8B),这表明HDAC3缺陷的MCA205细胞在体内具有远端抗肿瘤活性。为了确定CXCL10/CXCR3轴在HDAC3缺陷的MCA205细胞抗肿瘤活性中的作用,我们将WT, HDAC3+/−或HDAC3−/− MCA205纤维肉瘤细胞接种到左侧WT MCA205肿瘤小鼠的右侧,连续7天,然后静脉注射CXCR3抗体或IgG对照。CXCR3抗体恢复了MCA205的肿瘤生长(补充图S8C),提示Hdac3缺陷细胞的体内抗肿瘤活性依赖于CXCL10和CXCR3通路。

图7:CXCR3抗体阻断以免疫依赖的方式拯救Hdac3缺陷的MCA205肿瘤生长
(8) HDAC3与CXCL10在人HCC TME中的表达呈负相关
为了验证HDAC3和CXCL10在人体样本中的调节关系,我们收集HCC患者的恶性(n=86)和良性(n=41)组织,并进行TMAIHC评估HDAC3、CD8a、CXCL10和CXCR3的蛋白表达(补充表S1和S2)。癌组织中HDAC3的表达高于良性组织,而CXCL10的表达低于良性组织(补充图S9A和S9B)。此外,通过Kaplan-Meier分析根据HDAC3和CXCL10水平分类的不同亚组的临床结局,结果表明,HDAC3高表达或CXCL10低表达的HCC患者(cutoff=50%)的预后比HDAC3低表达或CXCL10高表达的HCC患者更差(补充图S9C)。为了观察HCC样本中HDAC3、CXCL10、CD8a和CXCR3之间的关系,我们进一步分析了HDAC3、CD8a、CXCL10和CXCR3水平之间的相关性。HDAC3蛋白水平不高,但与CD8a和CXCL10的表达呈显著负相关,CD8a的表达与CXCR3蛋白水平呈正相关(补充图S9D)。这些临床样本的结果表明,HDAC3可能调节CXCL10的表达,进而影响CD8+ T细胞在HCC TME中的浸润。
结论:我们证明了抑制HDAC3通过促进Cxcl9, 10, 11的表达来募集T细胞浸润到TME中,从而抑制肿瘤生长。这项工作可能有助于指导未来的癌症治疗,通过在肿瘤细胞中使用HDAC3-特异性抑制剂来治疗肿瘤细胞。
参考文献:Li, L., Hao, S., Gao, M., Liu, J., Xu, X., Huang, J., Cheng, G., & Yang, H. (2023). HDAC3 Inhibition Promotes Antitumor Immunity by Enhancing CXCL10-Mediated Chemotaxis and Recruiting of Immune Cells. Cancer immunology research, 11(5), 657–673. https://doi.org/10.1158/2326-6066.CIR-22-0317.