






ZBP1可预防mtDNA诱导的衰竭心脏的心肌炎症
栏目:最新研究动态
发布时间:2023-08-23
ZBP1(Z-DNA 结合蛋白1)是一种模式识别受体,可响应炎症细胞、成纤维细胞和内皮细胞中的mtDNA 而积极调节炎症......

心力衰竭(HF)的患病率和发病率正在上升,并被定义为一种全球大流行,影响全球约26万人。尽管治疗取得了重大进展,但HF的发病率和死亡率仍然很高。因此,需要对HF的病理生理学和分子机制有新的见解来开发新的治疗方法。线粒体DNA(mtDNA)诱导的心肌炎症与心脏重塑密切相关。ZBP1(Z-DNA 结合蛋白1)是一种模式识别受体,可响应炎症细胞、成纤维细胞和内皮细胞中的mtDNA 而积极调节炎症。然而,ZBP1在心肌炎症和心脏重塑中的作用尚不清楚。该研究发表于《Circulation Research》,IF:23.213。
技术路线:

主要研究结果:
1. mtDNA增加ZBP1并激活心肌细胞中的炎症信号传导
为了评估心肌细胞中ZBP1对mtDNA的表达,作者将1000ng / mL从大鼠肝脏中提取的mtDNA施用到心肌细胞中,通过检测mtDNA的一部分Cox1而不是核DNA的一部分At3来验证其纯度(图1A)。从mtDNA给药24小时后,作者发现ZBP1 mRNA和蛋白质水平增加(图1B和1C)随着IL-1β和IL-6 mRNA水平的增加(图1D)。
此外,作者研究了ZBP1和其他与炎症相关的蛋白质的mtDNA剂量依赖性反应,包括微管相关蛋白1A / 1B- LC3(轻链3),RIPK3,磷酸化的NF-κB p65亚基(Ser536)和NLRP3。虽然低剂量的mtDNA(100 ng/mL)增加了LC3-II,但没有增加ZBP1,但高剂量的mtDNA(1000 ng/mL)增加了LC3-II和ZBP1(图1E和1F)。重要的是,只有高剂量的mtDNA才能增加RIPK3,磷酸化的NF-κB,NLRP3和磷酸化TBK1的蛋白质水平,它们是炎症的主要介质(图1F)。此外,高剂量的mtDNA增加了IL-1β和IL-6的mRNA水平,而低剂量则没有(图1G)。这些数据表明,虽然低剂量的mtDNA通过自噬激活和处理,如LC3-II的增加所证明的那样,高剂量的mtDNA不仅诱导自噬,而且还增加ZBP1和其他炎症相关蛋白,导致心肌炎症。
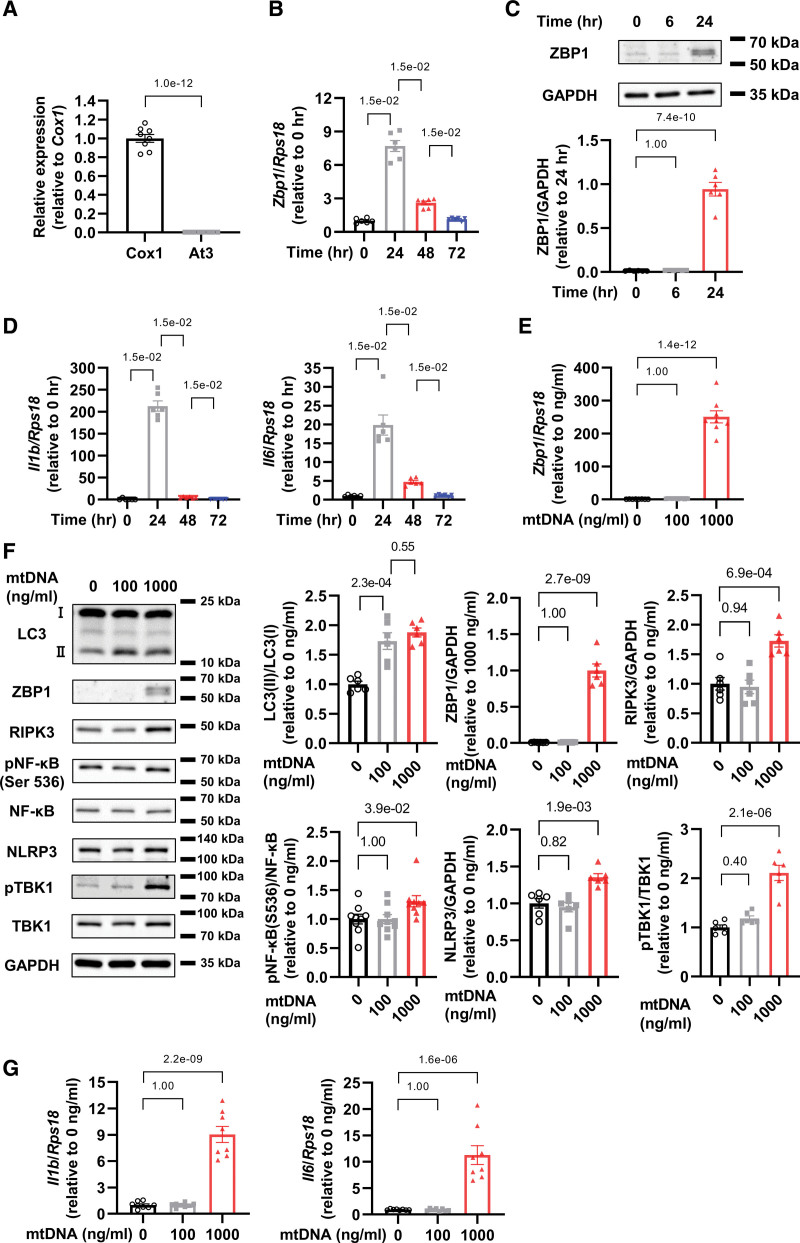
图1 mtDNA增加ZBP1并激活心肌细胞中的炎症信号传导
2. ZBP1敲低加剧了用mtDNA处理的心肌细胞中炎症细胞因子的增加
接下来,作者研究了ZBP1在心肌细胞中响应1000 ng / mL mtDNA的功能作用。小干扰RNA敲低有效抑制了ZBP1的mRNA和蛋白质水平(图2A和2B)。出乎意料的是,尽管ZBP-1已被报道介导炎症,但ZBP1敲低加剧了mtDNA诱导的RIPK3,磷酸化NF-κB和NLRP3的增加(图2B)。与这些结果一致,ZBP1敲低进一步增加了用mtDNA处理的心肌细胞中IL-1β和IL-6的mRNA水平(图2C)。TBK1是ZBP1的另一个下游靶标。尽管高剂量的mtDNA增加了TBK1的磷酸化(图1F),ZBP1敲低没有影响它(图2D)。这些发现表明,ZBP1负调节心肌细胞中的RIPK3,NF-κB和炎性细胞因子,但不是TBK1。

图2 ZBP1敲低会加剧用mtDNA处理的心肌细胞中炎性细胞因子的增加
3. 在mtDNA处理的心肌细胞中,敲低RIPK3可通过敲低ZBP1抵消NF-κB-NLRP3轴的增加
为了研究ZBP1和RIPK3之间的关系,并阐明RIPK3是否可以介导炎症作为ZBP1的下游靶标,作者进行了ZBP1和RIPK3的双重敲低实验。双敲低有效降低了这些蛋白质(图3A)。重要的是,RIPK3敲低减弱了mtDNA处理的心肌细胞中ZBP3敲低磷酸化NF-κB和NLRP1增加的恶化(图3A)。RIPK3敲低也降低了IL-1β和IL-6的mRNA水平(图3B)。这些发现表明RIPK3正调控NF-κB和NLRP3,ZBP1通过抑制RIPK3发挥抗炎作用。相反,mtDNA和ZBP1敲低都不影响细胞存活(图3C),表明ZBP1-RIPK3轴不能直接调节心肌细胞死亡。
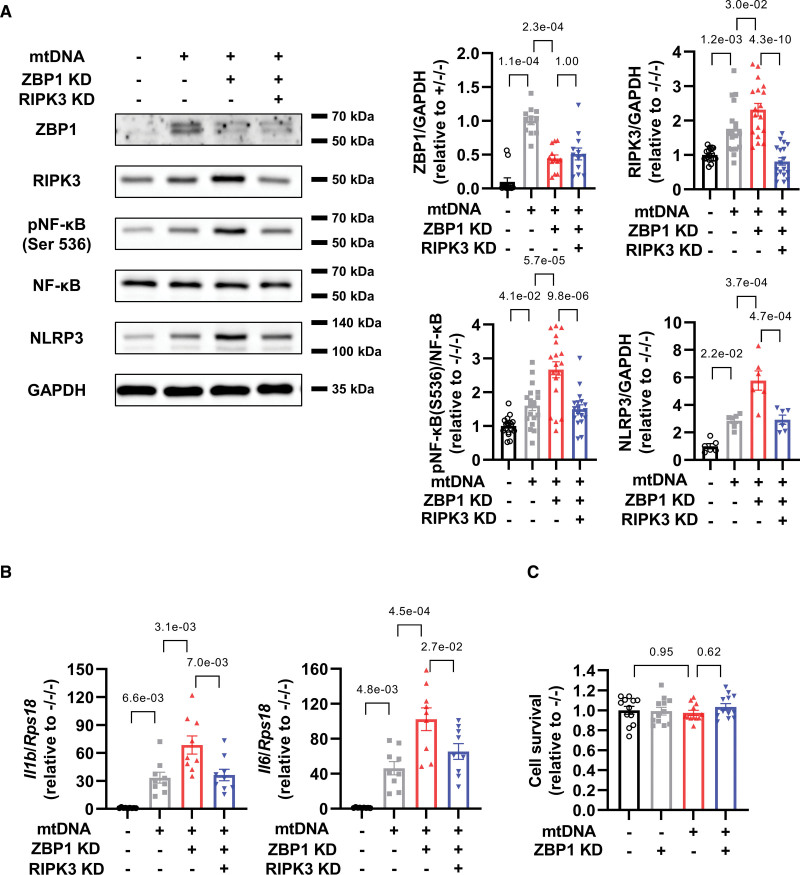
图3在mtDNA处理的心肌细胞中,敲低RIPK3可通过敲低ZBP1抵消NF-κB-NLRP3轴的增加
4. 在mtDNA处理的心肌细胞中,敲低NF-κB抑制了ZBP1敲低引起的NLRP3轴的增加
接下来,作者研究了NF-κB是否可以介导炎症作为ZBP1的下游靶标。作者发现NF-κB敲低减轻了mtDNA处理的心肌细胞中ZBP3敲低NLRP3增加的恶化,但不能减轻RIPK1的恶化(图4A)。重要的是,这些变化伴随着IL-1β和IL-6的mRNA水平的降低(图4B)。综上所述,这些数据表明ZBP1通过抑制NF-κB途径发挥抗炎作用。
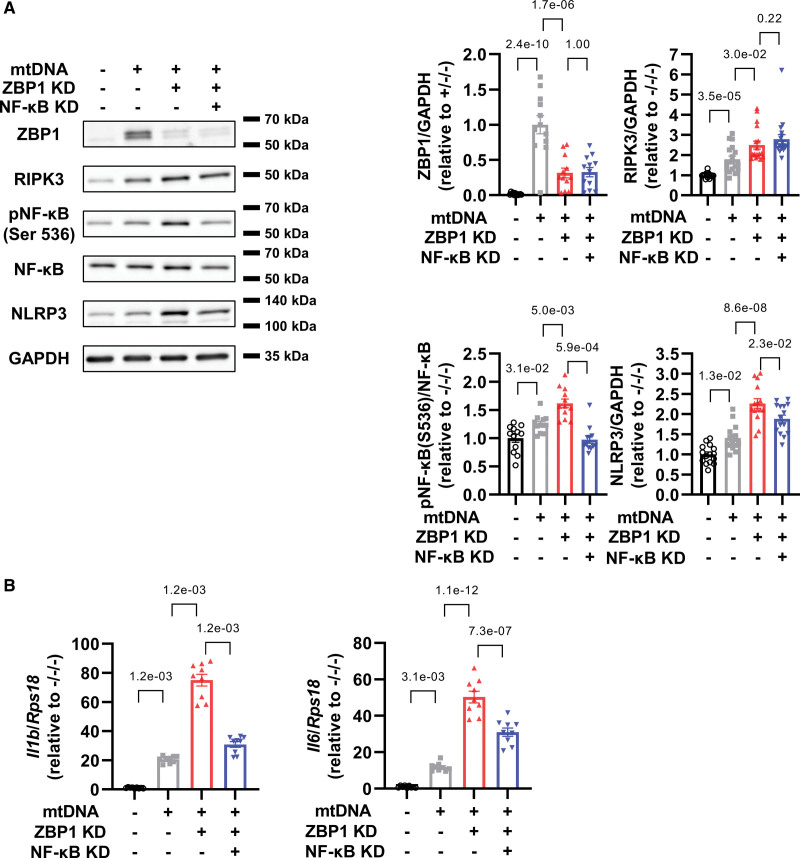
图4 在mtDNA处理的心肌细胞中,敲低NF-κB抑制了ZBP1敲低引起的NLRP3轴的增加
5. ZBP1负调节CpG-寡脱氧核苷酸处理的心肌细胞中的炎症反应
众所周知,ZBP1可以感知线粒体和核DNA。为了鉴定刺激心肌细胞中ZBP1的DNA类型,作者使用了模拟mtDNA的CpG-寡脱氧核苷酸和模拟核DNA的对照寡脱氧核苷酸。作者发现异硫氰酸荧光素(FITC)标记的CpG-寡脱氧核苷酸与mcherry标记的ZBP1共定位(图5A)。5CpG-寡脱氧核苷酸增加ZBP1 mRNA和蛋白质水平(图5B和5C)和ZBP1敲低进一步增加了用CpG-寡脱氧核苷酸处理的心肌细胞中IL-1β和IL-6的mRNA水平(图5D)。这些发现表明,ZBP1可以感知心肌细胞中的线粒体和核DNA,并负向调节炎症以响应它们。
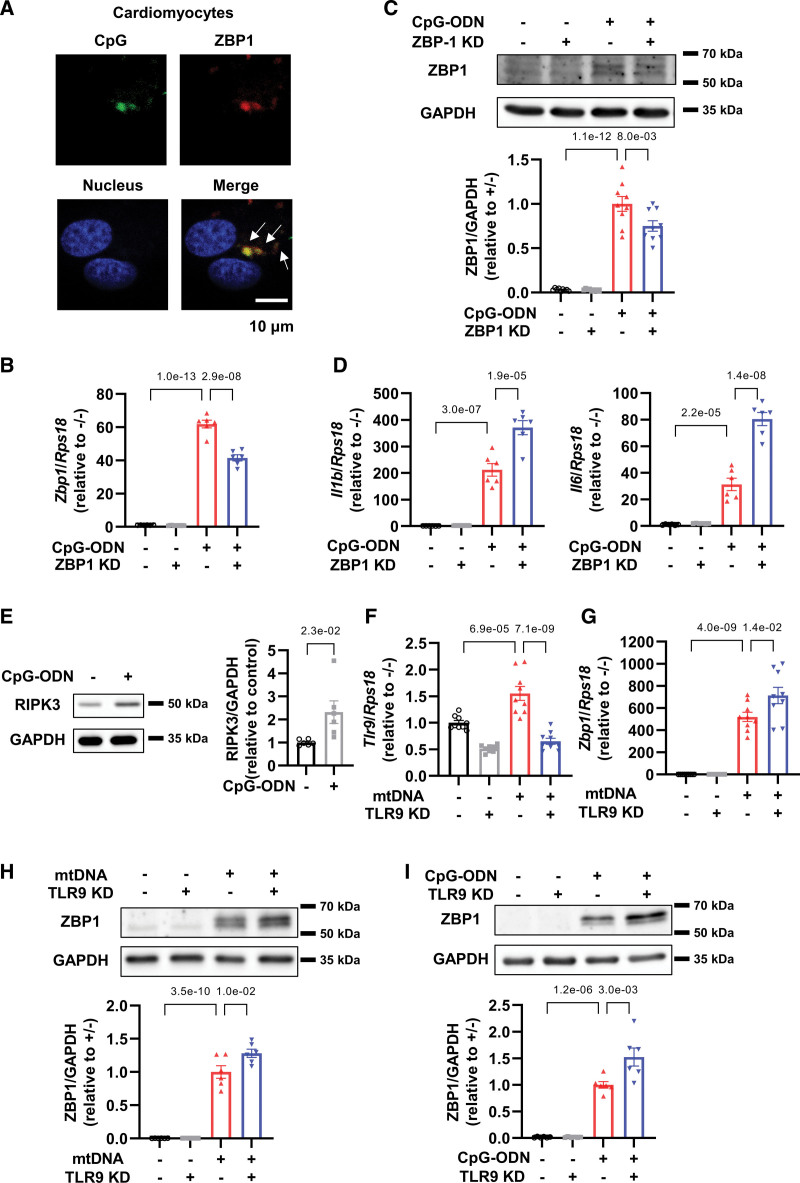
图5 ZBP1和CpG-ODN的相互作用参与ZBP1在CpG-ODN处理的心肌细胞中的抗炎作用
6. TLR9 敲低通过抑制 RIPK3、NF-κB 和 NLRP3 来改善 mtDNA 或 CpG-寡脱氧核苷酸处理的心肌细胞中的炎症反应
接下来,作者试图研究mtDNA处理的心肌细胞中TLR9,ZBP1和RIPK3之间的关系。CpG-寡脱氧核苷酸,也称为TLR9刺激剂,被发现增加心肌细胞中的RIPK3表达(图5E),表明RIPK3是TLR9的下游。作者证实TLR9敲低有效地降低了其mRNA水平(图5F)。此外,作者发现TLR9敲低增强了mtDNA诱导的心肌细胞中ZBP1 mRNA和蛋白质水平的增加(图5G和5H)和加剧的CpG-寡脱氧核苷酸诱导的ZBP1增加(图5I)。这些数据表明ZBP1阻碍了mtDNA和TLR9之间的相互作用,从而预防了下游炎症信号传导。
7. 心肌梗死后衰竭心脏的ZBP1和胞质mtDNA增加
为了评估ZBP1在衰竭心脏中的作用,作者在小鼠中进行了MI的体内实验。作者在MI模型中验证了梗死。作者发现,心肌梗死术后1天,心肌梗死后心脏非梗死区域的ZBP3蛋白水平升高(图6A)。接下来,作者分析了MI模型中胞质部分的DNA水平。作者对GAPDH、线粒体标志物COX4和自噬标志物LC3进行了免疫印迹验证了胞质组分的纯度(图6B)。Dloop是mtDNA的一种成分,在细胞溶质部分中在对照心脏中大量检测到,并在MI后心脏中增加(图6B)。相反,核DNA的组成部分Tert和B2m在对照和心肌梗死后心脏中均略有检测到(图6B)。总体而言,这些数据表明ZBP1与衰竭心脏中胞质mtDNA的增加有关。

图6 ZBP1敲除通过 RIPK3-NF-κB-NLRP3途径增强衰竭心脏的心肌炎症
8. ZBP1 敲除加剧心功能不全和心肌梗死后重塑
ZBP1基因敲除小鼠在心肌梗死后左心室(LV)舒张和收缩压直径增加,左心室射血分数降低,心脏衰竭部分缩短(图7A)。ZBP1敲除也加剧了全心重和左心室体重的增加(图7B)。在病理分析中,ZBP1敲除增强了衰竭心脏的胶原蛋白体积,这是心脏纤维化的指标,但不是横截面积,心肌细胞肥大的指数(图7C)。这些数据表明,ZBP1 敲除会加剧心肌梗死后的心功能不全和重塑。
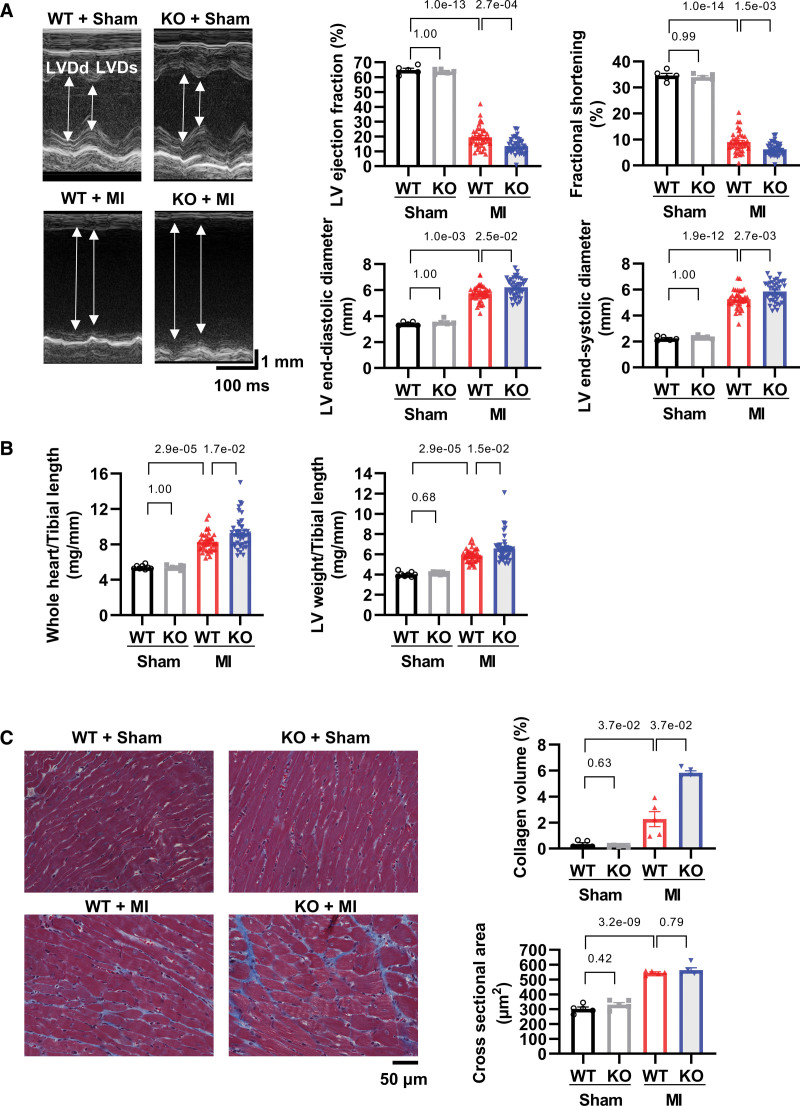
图7 ZBP1敲除会加剧心肌梗死后的心功能障碍和重塑
结论:
ZBP1作为RIPK3-NF-κB通路的心肌炎症内源性抑制因子,在心脏重塑中起保护作用。旨在通过ZBP1干扰心肌炎症的治疗策略可能有助于预防心脏重塑和衰竭。
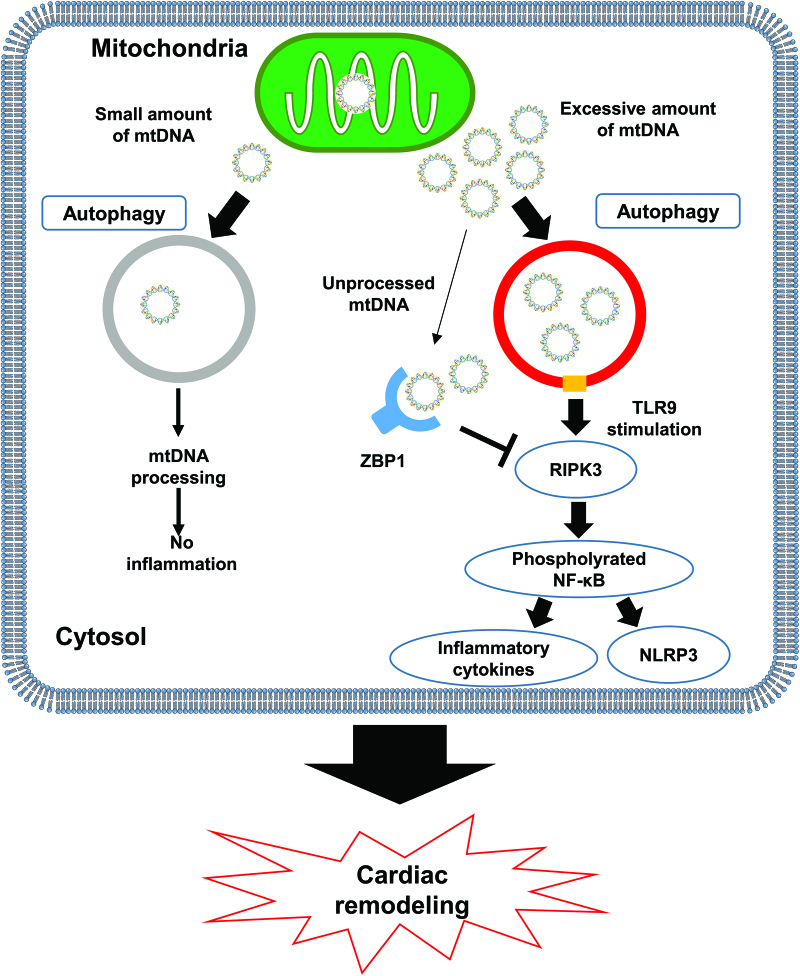
示意图:
ZBP1在心肌炎症和重塑中的作用示意图
实验方法:
细实时聚合酶链反应,免疫印迹,细胞活力检测,动物实验
参考文献:
Enzan N, Matsushima S, Ikeda S, Okabe K, Ishikita A, Yamamoto T, et al. ZBP1 Protects Against mtDNA-Induced Myocardial Inflammation in Failing Hearts. Circ Res. 2023 Apr 28;132(9):1110-1126.