






肝细胞癌的一区文章——分泌的蛋白酶PRSS35通过使CXCL2介导的中性粒细胞胞外陷阱来抑制肝细胞癌
肝细胞主要通过分泌调节细胞增殖、代谢和细胞间通讯的蛋白质发挥作用。在肝细胞癌(hepatocellular carcinoma,HCC)的进展过程中,肝细胞分泌蛋白质组作为肿瘤发生的结果和致病因素发生动态变化,尽管分泌蛋白质在这一过程中的全部功能尚不清楚。在这里,作者证明了分泌的伪丝氨酸蛋白酶PRSS35在HCC中作为肿瘤抑制因子发挥作用。在机制上,作者证明了有活性的PRSS35是通过前蛋白转化酶切割加工的。活化的PRSS35通过靶向切割串联赖氨酸(KK)识别基序抑制CXCL2的蛋白水平。因此,CXCL2降解减弱了中性粒细胞向肿瘤的募集和中性粒细胞胞外陷阱的形成,最终抑制了HCC的进展。这些发现拓展了作者对肝细胞分泌组在肿瘤发生发展中作用的理解,同时为PRRS35作为治疗靶点或诊断生物标志物的临床转化提供了依据。本研究与2023年3月发表于期刊《Nature Communications》上,IF:16.6。
技术路线

主要研究结果
1、PRSS35蛋白丰度在HCC分泌组中降低
为确定与HCC发生和进展相关的潜在因素,作者对HCC分泌组进行了无标记蛋白质组学分析,并比较了条件培养基中分泌的人PLC肝癌细胞和人THLE3肝细胞的蛋白质谱。作者发现,与THLE3细胞相比,有236种分泌蛋白在PLC细胞培养的条件培养基中显示出不同的丰度。PRSS35是PLC分泌组中下调最显著的蛋白(图1a)。使用识别蛋白N端的抗体进行Western blot(WB)分析PRSS35表明,相对于其在THLE3细胞中的积累,PLC、HepG2和Hep3B肝癌细胞内和细胞外PRSS35蛋白水平均显著降低(图1b)。有趣的是,富集在培养基中的PRSS35(SF-PRSS35)的分子量远低于从细胞裂解液中分离的全长PRSS35(FL-PRSS35)(图1b)。进一步利用针对PRSS35(即根据各自的抗原序列分别命名为N-PRSS35、M-PRSS35和C-PRSS35)不同肽段区域的抗体进行WB分析发现,在过表达PRSS35的PLC细胞培养基中富集到多个PRSS35蛋白短片段(图1c)。质谱分析(MS)也证实,SDS-PAGE检测到的较短波段确实包含了几个PRSS35肽(图1d)。总的来说,这些结果确定了PRSS35是一种分泌蛋白,在HCC细胞中丰度明显较低,可能是由于裂解成多个片段。
随后,作者发现与正常受试者相比,HCC患者血清中不同截短型PRSS35的水平显著降低(图1e),而全长PRSS35的水平无显著变化。为进一步定量患者血清中的PRSS35水平,作者使用针对N末端区域的两种抗体开发了定制的PRSS35 ELISA试剂盒。ELISA分析149例HCC患者和73例正常人血清,发现HCC患者血清PRSS35水平显著低于正常人(图1f)。这些结果表明分泌型PRSS35蛋白可以作为HCC患者潜在的预后生物标志物。此外,作者通过JASPAR数据库预测可能调控PRSS35的转录因子,发现HNF4A是最有潜力的候选转录因子。此外,作者观察到在HepG2细胞中敲低HNF4A后,PRSS35蛋白和mRNA水平均下调,而过表达HNF4A后,PRSS35蛋白和mRNA水平均上调。作者的数据表明HNF4A在肝癌中调控PRSS35的转录。
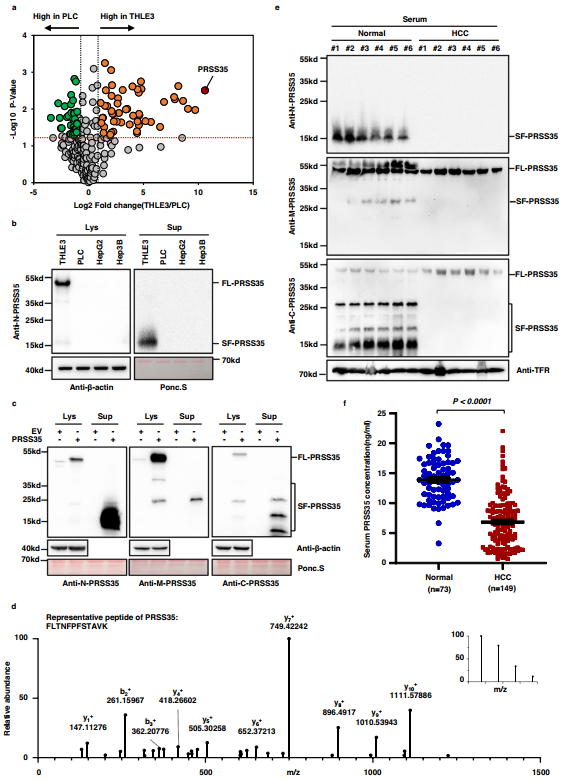
图1 PRSS35是一种分泌蛋白,在HCC患者中减少
2、PRSS35是一种活性蛋白酶
PRSS35被注释为无活性的假丝氨酸蛋白酶,因为在定义该酶家族的典型丝氨酸活性位点上存在苏氨酸。然而,最近的研究表明,它的功能与卵母细胞成熟、受精和胚胎发育或肾小管间质炎症有关,甚至调节伤口诱导的皮肤肿瘤发生。作者观察到PRSS35降低与HCC之间的关联,因此作者推测这种“假蛋白酶”可能表现出与活性丝氨酸蛋白酶相似的功能。为检验这种可能性,作者首先检测了PRSS35蛋白酶对β-酪蛋白的活性,β-酪蛋白是许多丝氨酸蛋白酶的广谱底物。为此,作者从大肠杆菌中纯化带有融合His标签的全长PRSS35(FL-PRSS35),然后与β-酪蛋白孵育。作者观察到在纯化的FL-PRSS35存在下,β-酪蛋白被完全切割,表明PRSS35是一种活性蛋白酶(图2a,左图)。由于已知许多蛋白酶在发挥作用之前会被前蛋白转化酶(proprotein converter,PC)切割而活化,因此作者在体外反应体系中对FL-PRSS35进行了WB分析。该分析揭示PRSS35存在多个截短形式,表明FL-PRSS35功能可能需要PC裂解成短的、有活性的形式(图2a,右侧面板)。
此外,作者通过对β-酪蛋白全长和裂解肽序列的LC-MS分析,确定了PRSS35在β -酪蛋白中的裂解位点。结果显示,全长β -酪蛋白的肽段在K44前立即含有氨基酸残基,但没有切割β -酪蛋白(图2b),说明K43和K44之间的位置是PRSS35的切割位点。
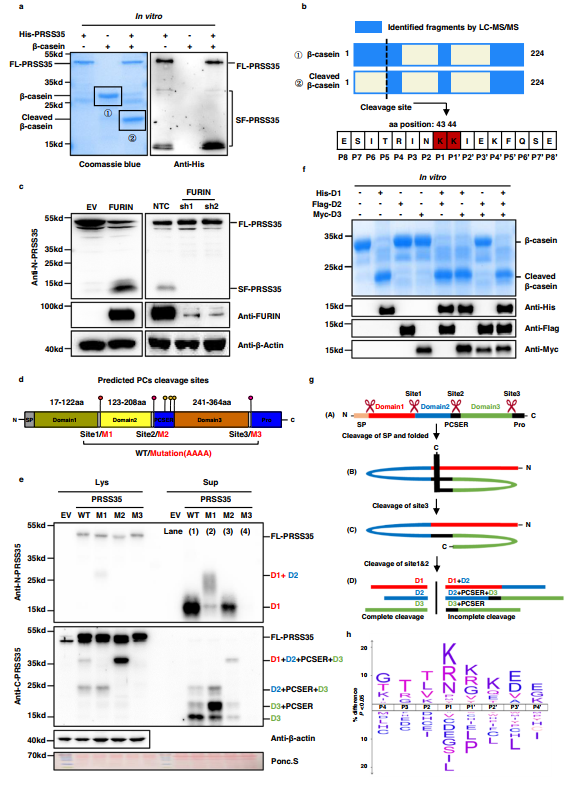
图2 PRSS35被FURIN激活后发挥蛋白酶的功能
3、PRSS35被蛋白转化酶激活
为研究PRSS35是否被PCs切割产生有活性的成熟体,作者研究了FURIN,一种广为人知且普遍存在的PC对PRSS35切割的影响。作者构建了FURIN过表达和敲低的PLC细胞系,然后用WB进行检测各系FL-PRSS35和SF-PRSS35水平。这些实验表明,在FURIN过表达的情况下,FL-PRSS35水平降低,SF-PRSS35水平升高,而在FURIN敲低的细胞系中则观察到相反的效果(图2c),表明PRSS35被FURIN和潜在的其他PCs切割。对PRSS35蛋白序列进行生物信息学分析,发现人PRSS35至少存在6个潜在的PC切割位点。作者进一步预测来自其他物种的PRSS35的PC剪切位点,并在UniProt数据库中找到了3个在所有物种中高度保守的剪切位点,包括小鼠和大鼠(图2d)。值得注意的是,这3个额外的预测切割位点仅存在于人类PRSS35的site2附近区域,作者将其命名为PC切割位点富集区域(PCSER;图2d)。
为确认PRSS35中预测的这三个高度保守的切割位点是真正的PC识别位点,作者分别在每个位点引入了非同义突变(AAAA)(图2d)。这些突变破坏了PRSS35的切割,每个PRSS35突变体在WB分析中表现出不同的特异性条带模式(图2e)。首先,位点3(M3)的突变导致了所有SF-PRSS35的消失,表明M3对于所有PRSS35的切割是必需的,这意味着该位点的切割是后续蛋白水解加工(图2e,泳道4与泳道1对比)所必需的。位点2(M2)突变导致由domain1(D1 , 12.4kd)或domain3(D3 , 14.2kd)组成的短型积累减少。此外,由结构域1、2、3和PCSER(即D1 + D2 + PCSER + D3)组成的中间片段增加了(图2e ,泳道3与泳道1对比),这表明在位点2的剪切促进了随后的PRSS35短形式的剪切。相反,位点1突变(M1)只抑制了导致domain1(D1 , 12.4kd)产生的剪切,并导致携带domain1和2(D1 + D2)(图2e ,泳道2与泳道1的比较)的中间片段的积累。这些发现表明作者预测的切割位点是PRSS35上真正的PC识别位点,这些位点的切割对于PRSS35的成熟至关重要。
为确定哪种形式的PRSS35具有活性蛋白酶功能,作者从大肠杆菌中纯化了His-D1、Flag-D2和Myc-D3融合蛋白片段。将这些纯化的蛋白片段分别与β -酪蛋白孵育,发现β-酪蛋白在体外中只有D1存在时才被切割(图2f),表明只有D1短形式具有活性。考虑到体外反应中呈现的短形式的分子量,作者的数据巩固切割β-酪蛋白的是(12.4 kD)的D1短体,而不是FL-PRSS35(图2a)。总之,这些结果表明PRSS35在被PC切割后发挥蛋白酶的作用,产生只含有D1的活性截短体。
为更好地了解这些预测的切割位点在PRSS35成熟过程中的作用模式和不同作用,作者进行了生物信息学分析,发现PRSS35的N端16个氨基酸很可能是信号肽。根据作者的WB数据,作者提出在N端的一个初始切割位点去除了FL-PRSS35(图2g,构象A)的信号肽(SP),导致PRSS35的折叠构象,其中位点3暴露在蛋白表面,位点1和2埋在蛋白内部(图2g,构象B)。PCs对位点3的切割改变了PRSS35的构象,暴露了位点1和位点2。然后,PCs可以被招募到邻近的PCSER中的位点2和位点1(图2g,构象C),最终导致PRSS35完全裂解为含有活性D1的短形式,以及含有D2、D3(图2g,构象D)的非活性片段。
为进一步鉴定PRSS35识别的切割基序,确定其靶底物,作者采用了高通量蛋白酶筛(HTPS)。研究分析切割窗口发现,两个相邻的赖氨酸氨基酸(KK)作为PRSS35识别的核心切割基序(图2h),这与作者在β -酪蛋白中鉴定到的切割位点一致(图2b)。此外,作者合成了3条含有预测切割基序的荧光肽段(图2h),发现His-D1肽段在体外中可以切割3条荧光肽段。这些发现证明PRSS35是一种由PCs裂解激活的胰蛋白酶样丝氨酸蛋白酶,其活化的短形式D1可以通过靶向KK识别位点降解底物。
4、PRSS35在体内抑制HCC的发展
作者的数据表明,PRSS35是一种在肝癌中被抑制的活性蛋白酶,这促使作者研究PRSS35与肿瘤发生的关系。作者发现,与EV对照组相比,过表达小鼠PRSS35(mPRSS35)显著抑制了Hepa1-6细胞在C57BL/6j小鼠中的生长(图3a)。然后作者通过流体动力学注射质粒YAP-5SA41建立自发性HCC小鼠模型。结果显示,mPRSS35过表达显著抑制小鼠肝癌的发生(图3b)。在YAP-5SA诱导的HCC小鼠模型中,作者观察到与WT小鼠相比,mPRSS35-KO小鼠发生肝癌的速度加快(图3c)。除了Hepa1-6小鼠HCC模型和YAP-5SA诱导的小鼠HCC模型外,作者还通过异种移植HCC模型证实了PRSS35在体内成瘤中的作用。作者发现人PRSS35(hPRSS35)过表达显著抑制Balb/C纯系裸鼠中HepG2肿瘤的生长(图3d)。这些结果表明mPRSS35有助于小鼠的肿瘤抑制,尽管其在体外缺乏对HCC细胞生长的影响。
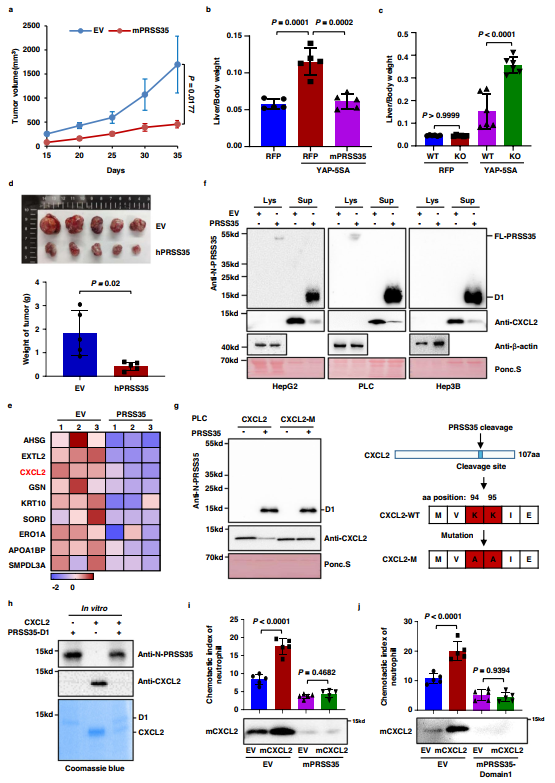
图3 PRSS35通过降解CXCL2抑制中性粒细胞迁移
5、CXCL2是PRSS35的底物
为探索微环境中是否有某些因素影响PRSS35,作者结合RNA测序进行了SILAC蛋白质组学分析,以鉴定在PRSS35过表达PLC细胞的培养基中积累减少但转录水平不降低的分泌蛋白。该分析确定了9种分泌蛋白作为PRSS35的潜在底物(图3e)。由于PRSS35的肿瘤抑制活性似乎依赖于体内微环境,鉴于CXCL2在肿瘤免疫微环境中作为趋化因子的作用,作者将研究重点放在了作为候选底物的CXCL2上。
WB分析表明过表达PRSS35显著降低了HepG2、PLC和Hep3B细胞中CXCL2的胞外水平(图3f)。此外,肽段序列分析表明CXCL2含有KK切割位点(图3g,右),该基序的突变阻断了PRSS35对CXCL2的切割(图3g,左),进一步支持CXCL2是PRSS35的底物。为排除PRSS35降低CXCL2蛋白水平的其他间接机制,将E. coli纯化的重组D1与E. coli纯化的CXCL2蛋白共孵育,导致CXCL2降解(图3h),证实CXCL2是PRSS35体外蛋白水解活性的直接底物。
由于CXCL2是趋化中性粒细胞的趋化因子,因此作者试图确定PRSS35降解CXCL2是否可以抑制中性粒细胞的募集。对中性粒细胞迁移的分析显示,从表达mCXCL2的细胞中收集的条件培养基增强了中性粒细胞的迁移,而过表达mPRSS35可以消除这种现象(图3i,上)。与这些研究结果一致,WB显示mPRSS35存在时,条件培养基中的mCXCL2蛋白显著降解(图3i,下)。对于PRSS35的活性D1形式也观察到类似的结果(图3j),从而证明mPRSS35介导的mCXCL2降解是中性粒细胞活性的促进因素。
5、PRSS35通过减少中性粒细胞向肿瘤的募集和减少NETs的形成来抑制HCC的进展
为研究PRSS35降解CXCL2在体内肿瘤进展中对中性粒细胞活性的影响,作者构建了稳定表达mPRSS35、mCXCL2和EV对照的Hepa1-6细胞系,并将这些细胞系分别接种到C57BL/6j小鼠皮下。结果表明,mCXCL2过表达促进了小鼠肿瘤的生长,而表达mPRSS35的小鼠表现出最低的肿瘤发生,有或无mCXCL2共表达(图4a)。肿瘤组织的WB分析证实,在mPRSS35存在的情况下,mCXCL2蛋白被耗竭(图4b)。进一步通过流式细胞术和IHC检测肿瘤组织发现,在过表达mCXCL2的小鼠中,中性粒细胞向肿瘤内的募集增强,而与mPRSS35共表达后,中性粒细胞向肿瘤内的募集减弱(图4c,e),表明mPRSS35介导的mCXCL2降解可以限制小鼠HCC组织中中性粒细胞的募集。
作者利用WB检测肿瘤组织中NETs形成的标志物瓜氨酸化组蛋白H3(H3Cit)的水平,发现其在过表达mCXCL2的小鼠中显著升高,但在过表达mPRSS35的小鼠中减弱(图4b)。这些结果提示PRSS35和CXCL2在NETs形成过程中诱导了相反的作用。此外,作者通过ELISA检测了荷瘤小鼠血清中H3Cit和另一个NETs形成的指标髓过氧化物酶- DNA(MPO-DNA)复合物。与mCXCL2促进H3Cit水平一致,作者发现mCXCL2过表达Hepa1-6细胞荷瘤小鼠血清中H3Cit和MPO-DNA复合物水平显著升高,而mPRSS35过表达Hepa1-6细胞荷瘤小鼠血清中H3Cit和MPO-DNA复合物水平在mCXCL2过表达和mCXCL2过表达荷瘤小鼠血清中均无显著变化(图4d)。
在YAP-SSA诱导的HCC模型小鼠中,与上述在转基因Hepa1-6诱导的HCC小鼠中得到的结果一致,作者发现与WT小鼠相比,mPRSS35-KO小鼠的肿瘤发展加快,而shRNA敲低mCXCL2(shmCXCL2)减弱了这种作用。此外,作者在荷瘤mPRSS35-KO小鼠的肿瘤组织和血清中观察到更高的中性粒细胞浸润和NETs形成,但在shmCXCL2的存在下被抑制(图4g)。这些数据表明PRSS35可以通过降解CXCL2抑制肿瘤中性粒细胞浸润和NETs形成。
为进一步研究中性粒细胞和NETs是否参与PRSS35介导的抑癌作用,将稳定表达空载体(EV)或Flag-mPRSS35的Hepa1-6细胞注射到C57BL/ 6J小鼠皮下。在细胞注射后第9天,作者将中性粒细胞中和Ly6G抗体或IgG对照以3天间隔注射到肿瘤和周围皮下组织中。再次,与EV对照组相比,mPRSS35表达显著抑制肿瘤生长,而在有或没有Flag-mPRSS35过表达的肿瘤中,Ly6G处理导致类似的效果(图4h)。与此一致,mPRSS35表达显著抑制中性粒细胞肿瘤浸润和NETs形成,Ly6G处理在Flag-mPRSS35过表达和不表达的肿瘤中产生类似的效果(图4i-k)。结果表明,PRSS35通过抑制CXCL2介导的中性粒细胞募集和NETs形成来抑制HCC进展。
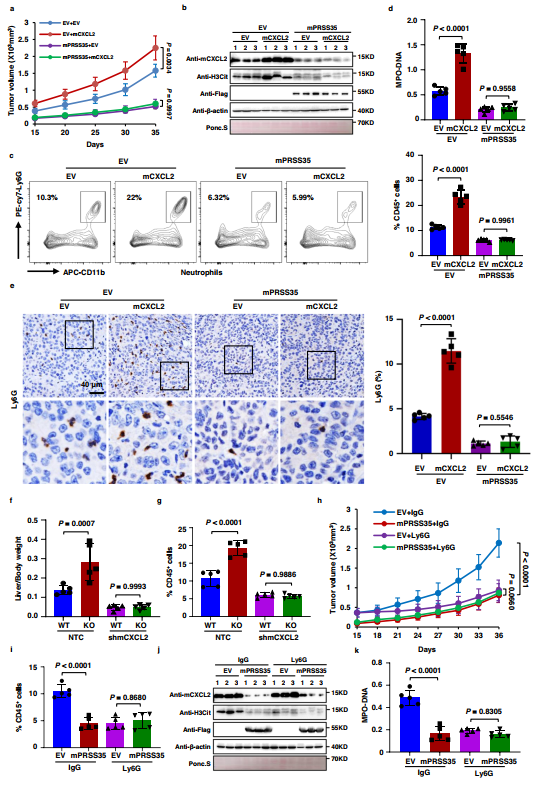
图4 PRSS35通过抑制cxcl2介导的中性粒细胞NETs形成来抑制HCC的发展
实验方法
人HepG2、Hep3B、PLC、Hepa1-6和HEK293T细胞培养,Western blot,ELISA法检测血清PRSS35,凝胶内质谱,肝癌细胞系的生长曲线,高通量蛋白酶筛选,C57BL/6J小鼠,Balb/c nude老鼠,异种移植瘤模型,流式细胞术,临床数据分析,血清MPO-DNA检测,中心粒细胞隔离,中心粒细胞迁移检测,PRSS35体外底物测定
参考文献
Wang T, Zhou Y, Zhou Z, Zhang P, Yan R, Sun L, Ma W, Zhang T, Shen S, Liu H, Lu H, Ye L, Feng J, Chen Z, Zhong X, Wu G, Cai Y, Jia W, Gao P, Zhang H.(2023) Secreted protease PRSS35 suppresses hepatocellular carcinoma by disabling CXCL2-mediated neutrophil extracellular traps. Nat Commun.;14(1):1513. doi: 10.1038/s41467-023-37227-z.