






沙门氏菌效应物SopF调节肠上皮细胞泛凋亡以加重全身感染

S. Typhimurium是引起广泛宿主腹泻的最常见食源性致病菌之一。抗生素耐药S. Typhimurium的出现是一个重大的公共卫生问题。另一方面,S. Typhimurium也是研究宿主-细菌相互作用机制的一个有用的模式生物研究细菌与宿主免疫反应相互作用的新模式,将为鼠伤寒沙门菌感染及其他感染性疾病的防治提供新思路。肠上皮细胞(IECs)本身不仅是保护黏膜免受病原体入侵的机械屏障,而且还协调一些固有免疫防御。泛凋亡(PANptosis)是一种以细胞焦亡、凋亡和(或)坏死性凋亡为主要特征的炎性程序性细胞死亡(PCD)。目前已知IECs的泛凋亡在宿主抵抗S. Typhimurium感染的过程中起关键作用。细胞焦亡和凋亡驱动被感染的IECs排出,以限制S. Typhimurium的复制。死亡和被挤压的IECs引起邻近上皮的收缩,以维持肠屏障的完整性。相反,IECs的坏死性凋亡可导致肠屏障破坏,并促进S. Typhimurium扩散至固有层。目前尚不清楚S. Typhimurium如何克服IECs的泛凋亡以维持生存。该研究发表在《Gut Microbes》,IF:12.2。
技术路线
示意图

主要研究结果
1. 鼠伤寒沙门菌效应蛋白SopF通过抑制肠道炎症加重全身感染
为了研究SopF在S. Typhimurium发病机制中的潜在作用,作者首先监测了在感染过程中的死亡率和体重变化。链霉素预处理小鼠经口感染STM-WT、STM-ΔsopF或STM-ΔsopF/ psopF。小鼠在4 dpi时出现多种濒死状态,灌胃STM-WT的小鼠在第2天死于感染。在同一时间内,70%的STM-ΔsopF-infected小鼠仍然存活。此外,感染STM-ΔSopF/pSopF的小鼠在6天内死于感染(图1a)。随着感染的进行,作者观察到小鼠的体重逐渐下降,但3组之间没有任何显著差异(图1b)。SopF在S. Typhimurium的早期固有免疫应答中发挥重要作用,并影响感染的结局。与之前的研究一致,与感染STM的小鼠相比,感染携带SopF的S. Typhimurium的小鼠在48 hpi时其肝脏和脾脏中的细菌负荷约为STM-ΔsopF的2倍,在120 hpi时观察到细菌负荷显著增加(图1c-d)。这些数据证实,SopF是S. Typhimurium在肠外器官播散所必需的。
肠道炎症反应是宿主与病原体相互作用的重要机制接下来,作者关注SopF对S. Typhimurium诱导的肠道炎症的影响。在感染STM-ΔSopF的小鼠中,在48 hpi时观察到的结肠长度收缩大于感染STM-WT和STM-ΔSopF/pSopF的小鼠(图1e-f)。所有鼠伤寒沙门菌感染的小鼠均出现进行性病理改变,包括肿胀和炎性细胞浸润至固有层和黏膜下层。重要的是,SopF缺乏加重了S. Typhimurium诱导的盲肠组织病理学损伤,包括48 hpi时的IECs排出和120 hpi时的黏膜组织中IECs丢失(图1g-h)。为了进一步表征SopF引起的肠道炎症反应,作者分析了与宿主防御细胞内病原体密切相关的炎症细胞因子水平如图1i-k所示,随着感染时间的延长,盲肠中Il-6、Tnf-α和Il-1β的转录水平被诱导的最明显,在感染后120 hpi时,与STM- wt和STM-ΔSopF /pSopF感染小鼠相比,STM-ΔSopF感染小鼠盲肠中细胞因子的表达显著增加。综上所述,这些结果表明SopF抑制肠道炎症,从而加重S. Typhimurium感染的全身性感染。
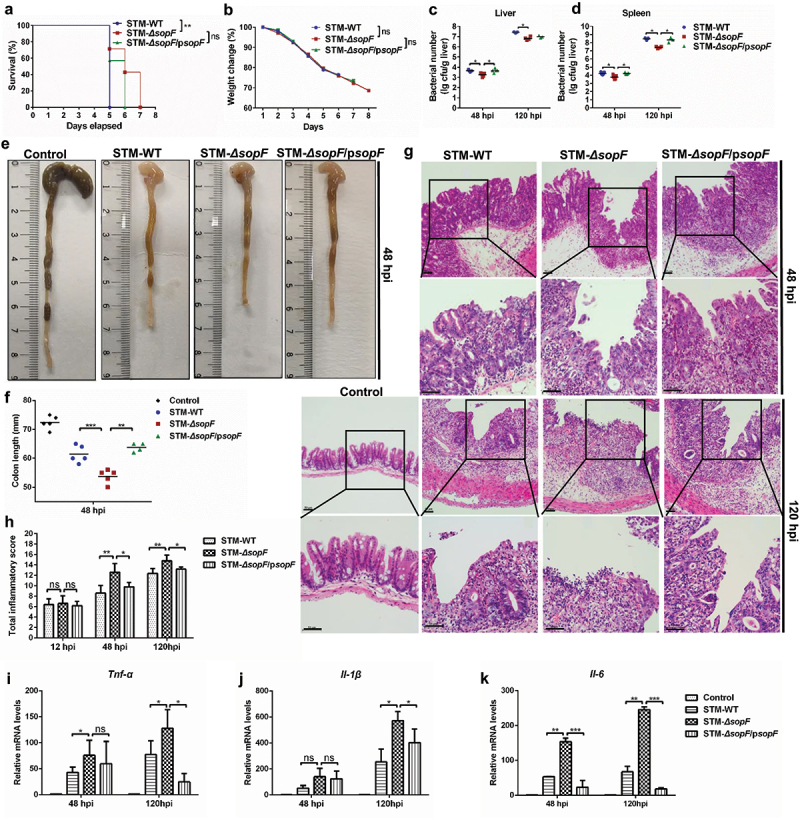
图1 SopF抑制肠道炎症,加重全身感染
2. S. Typhimurium效应因子SopF通过限制IECs的脱落促进细菌播散
IECs在将管腔中的微生物群落从无菌系统环境中分离出来方面起着重要作用。受感染的IECs排出限制了病原体的上皮内增殖,是机体抵御肠道病原体感染的一般防御机制。为了确定由SopF介导的S. Typhimurium与IECs之间的相互作用,作者采用荧光显微镜观察IECs屏障的完整性和S. Typhimurium的分布。在120 hpi时,作者观察到在感染STM-ΔsopF的小鼠的黏膜组织中,总IECs数量显著丢失,每10倍视野的上皮间隙较多(图2a-d)。作者进一步用透射电子显微镜观察盲肠的超微结构(图2b)。在感染早期(12 hpi)肠腔内可见细菌(图2b-c)。为了评估IECs排出与细菌播散开始之间可能的相关性,作者通过监测感染的时间进程来扩展这些研究。感染后48 h,小鼠盲肠出现刷状边缘结构紊乱、微绒毛减少、变短等异常。相比之下,感染携带SopF的S. Typhimurium的小鼠在48 hpi时的IECs中出现轻微损伤,但在120 hpi时黏膜下层的细菌载量显著较高(图2b)。抗S. Typhimurium- LPS染色显示,在感染STM的小鼠中,盲肠的细菌定植在12 hpi时增加(图2c)。在感染后期(120 hpi),携带SopF的S. Typhimurium主要定植在黏膜下层,而STM-ΔsopF仍在黏膜中(图2c-e)。综上所述,这些结果表明SopF限制IECs的排出以促进细菌的传播。

图2 SopF限制IECs的移位以促进细菌传播
3. 鼠伤寒沙门菌效应蛋白SopF在宿主防御鼠伤寒沙门菌感染过程中调节肠上皮细胞的泛凋亡
虽然上皮细胞主要被认为是一个机械屏障,但研究表明,IECs的细胞命运允许它们对抗病原体。作者研究了SopF在S. Typhimurium感染后IECs细胞死亡中的作用。在携带SopF的S. Typhimurium感染的人结肠癌Caco-2细胞和人正常结肠上皮NCM460细胞中,发现乳酸脱氢酶(LDH)释放减少,揭示了SopF抑制了IECs的细胞死亡(图3a-b)。最近的证据表明,在全凋亡方面,病原体和宿主之间存在相互作用。为了评估SopF对IECs凋亡的影响,作者从S. Typhimurium感染的小鼠盲肠中分离IECs。蛋白质印迹分析显示,STM-ΔsopF-infected小鼠体内GSDMD-NT片段蛋白水平较高,可形成膜孔诱导细胞焦亡。被Caspase-3裂解的上皮源性GSDME也参与肠道炎症的发病机制32因此,作者在STM-ΔsopF-infected小鼠中发现了稳健的GSDME激活(图3c-d)。上述结果提示SopF抑制IECs细胞焦亡。除了细胞焦亡,作者发现在感染STM-ΔsopF的小鼠中,凋亡执行体caspase(caspase-3)的切割增加(图3e-f)。焦亡和凋亡的IECs尖化以维持上皮屏障的完整性,而SopF阻止感染的IECs被清除。IECs的坏死性凋亡也会导致上皮屏障功能障碍,从而促进S. Typhimurium的传播。接下来,作者研究了SopF是否也参与了IECs的程序性坏死。正如预期的那样,在携带SopF的S. Typhimurium感染的小鼠中,在12 hpi时观察到混合谱系激酶结构域样蛋白(MLKL)的强劲磷酸化,这表明SopF促进IECs的坏死性凋亡,从而使S. Typhimurium播散到固有层甚至肠外器官(图3g-h)。在Caco-2细胞(图3i-n)中显示类似结果。综上所述,这些数据表明SopF通过抑制细胞凋亡和焦亡而促进IECs的坏死性凋亡来调节泛凋亡,这可能与S. Typhimurium的系统性感染有关。
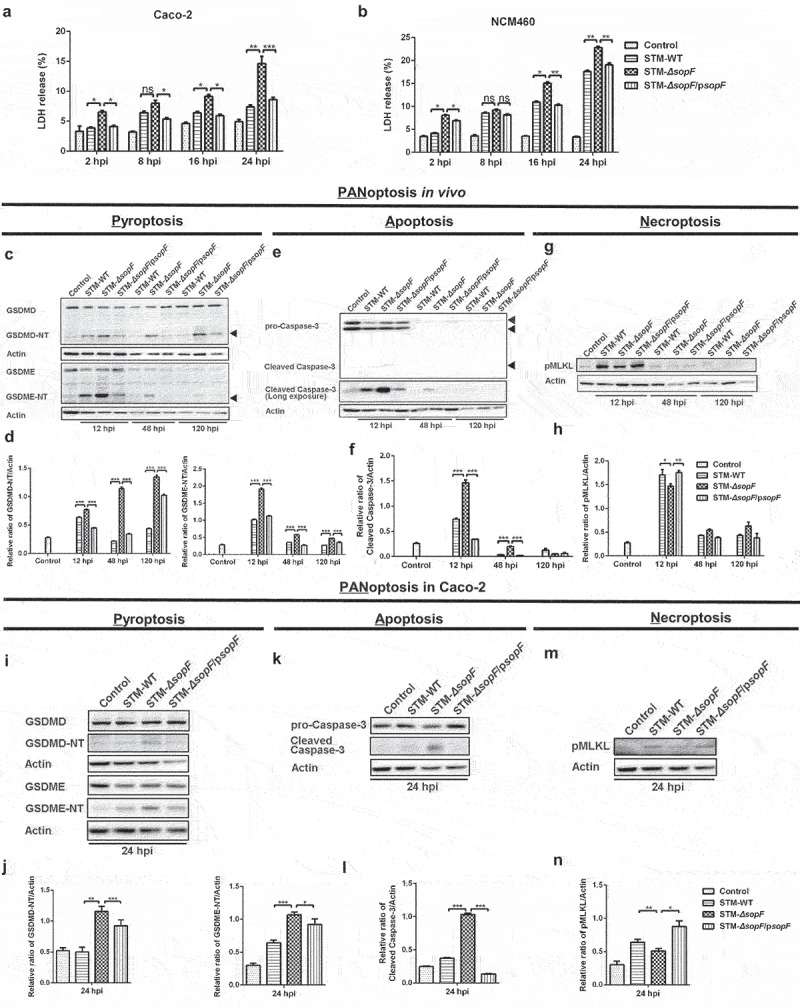
图3 SopF调节肠上皮细胞的泛凋亡以防御宿主感染S. Typhimurium
4. 鼠伤寒沙门菌效应蛋白SopF可抑制细胞凋亡的分子开关Caspase-8的激活
Caspase-8通过细胞表面死亡受体连接和寡聚化激活,是细胞凋亡、坏死性凋亡和细胞焦亡的分子开关。此外,Caspase-8调控上皮细胞PCD,防止了S. Typhimurium感染时上皮细胞屏障功能障碍为了了解SopF对Caspase-8的潜在影响,作者从S. Typhimurium感染的小鼠盲肠分离IECs进行了检测。与野生型或补充突变株感染的小鼠相比,缺乏SopF的沙门菌感染的小鼠在12和48 hpi时,Caspase-8的切割增加(图4a-b)。相应的,在Caco-2细胞和NCM460细胞中,SopF的缺失导致Caspase-8在24 hpi时的切割增加(图4c-f)。总之,这些数据表明,SopF通过抑制Caspase-8的激活来调节IECs的泛凋亡。
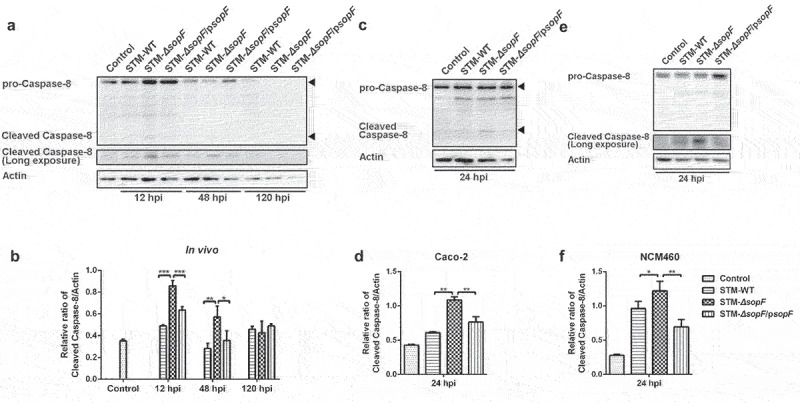
图4 SopF抑制Caspase-8的活化,Caspase-8是泛凋亡的分子开关
5. 在SopF介导的S. Typhimurium感染中,PDK1-RSK信号对Caspase-8阻断是必需的
Caspase-8可能作为调控泛凋亡的守门人。Yang等人之前的工作表明,3-磷脂酰肌醇依赖性蛋白激酶1(PDK1)-核糖体S6激酶(RSK)信号是减少Caspase-8阻断的内在机制26值得注意的是,SopF与宿主细胞膜的磷脂酰肌醇结合,从而促进新生沙门菌空泡(SCV)的稳定9因此,作者假设PDK1-RSK信号通路参与了SopF介导的Caspase-8激活的阻断。蛋白质印迹分析显示,携带SopF的S. Typhimurium感染的小鼠中,PDK1和磷酸化RSK的蛋白水平可能高于STM-ΔSopF-infected组(图5a-b),表明SopF可以激活PDK1-RSK通路,从而取消Caspase-8的激活。在293 T细胞中表达的His或His-SopF被免疫沉淀,并评估其与内源性PDK1相互作用的能力。结果显示,在His-SopF免疫沉淀样品中,未检测到PDK1(图5c)。然后,作者检测了在感染的Caco-2细胞中,被S. Typhimurium易位的SopF:His是否与内源性PDK1相互作用。构建携带空pBAD的STM-ΔsopF(STM-ΔsopF/pBAD)或带His标签的SopF表达的pBAD(STM-ΔsopF/p sopF: His)构建感染模型。在SopF中也未检测到PDK1:His免疫沉淀样本在感染早期和晚期均未检测到PDK1,这表明SopF不能直接与PDK1相互作用(图5d)。
AR-12和BI-D1870分别是PDK1和RSK的ATP拮抗剂。PDK1抑制剂AR-12可能消除了24 hpi时RSK磷酸化的增加,但逆转了SopF引起的Caspase-8活化的降低。此外,在S. Typhimurium感染的Caco-2细胞中,RSK抑制剂BI-D1870对Caspase-8也有相同的作用(图5e-g)。结果表明,SopF通过PDK1-RSK信号通路抑制Caspase-8的切割。鉴于Caspase-8在调节泛凋亡中起核心作用,作者进一步用AR-12或BI-D1870预处理Caco-2细胞。蛋白质印迹分析显示,AR-12和BI-D1870处理后,GSDMD-NT、GSDME-NT和cleaved Caspase-3蛋白水平升高,MLKL磷酸化水平降低。经抑制剂治疗后,3组感染组间差异无统计学意义。数据表明,AR-12和BI-D1870均恢复了停止的细胞焦亡和凋亡以及提示的坏死性凋亡(图5f-h)。
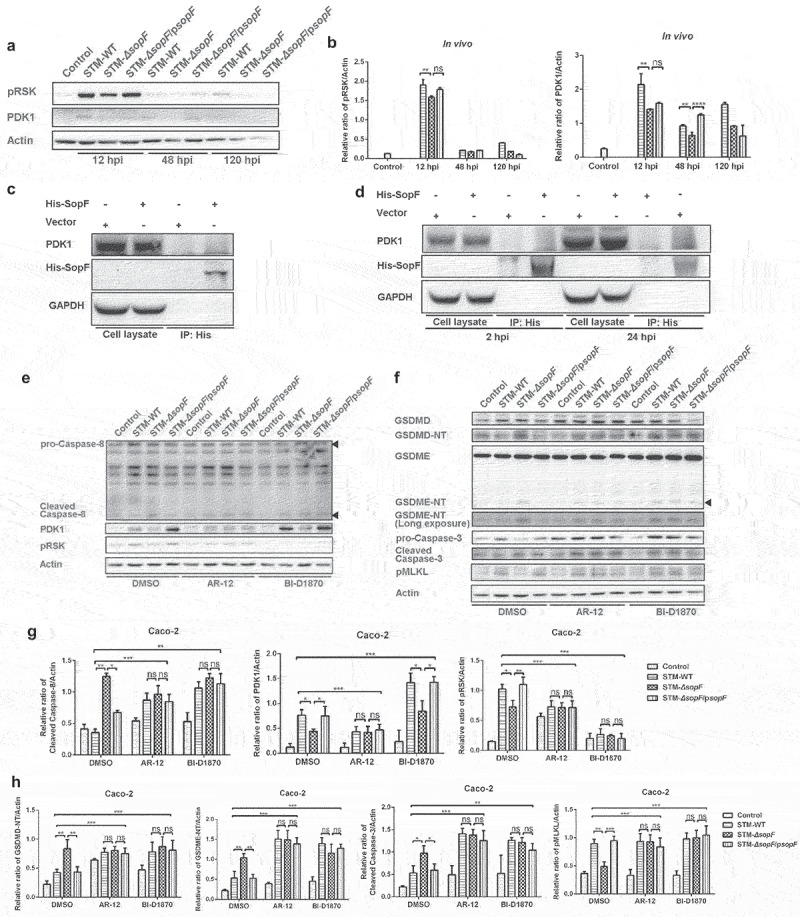
图5 在SopF介导的S. Typhimurium感染中,PDK1-RSK信号对Caspase-8阻断是必需的
结论
综上所述,作者确定了SopF在调节IECs 泛凋亡以加重S. Typhimurium播散方面的一种新功能。在这一范式中,PIP结合效应因子SopF通过PDK1-RSK信号通路阻断了Caspase-8的激活。由于Caspase-8在PCD中的重要作用,Caspase-8位点作为调控泛凋亡的十字路口。抑制Caspase-8可减少细胞焦亡和凋亡,增加程序性坏死。阻断PDK1或RSK的潜在治疗靶点逆转了对Caspase-8的阻断和随后的IECs 泛凋亡。SopF抑制的细胞焦亡和凋亡阻碍了感染IECs的清除,而SopF诱导的程序性坏死促进了S. Typhimurium的内化,从而促进细菌的播散。这些发现揭示了SopF调控IECs细胞命运的新机制,这可能为控制S. Typhimurium感染和其他相应疾病提供有吸引力的治疗策略。
实验方法
乳酸脱氢酶(LDH)释放试验,转染实验,免疫沉淀,蛋白质印迹分析,定量聚合酶链反应
参考文献
Yuan H, Zhou L, Chen Y, You J, Hu H, Li Y, Huang R, Wu S. Salmonella effector SopF regulates PANoptosis of intestinal epithelial cells to aggravate systemic infection. Gut Microbes. 2023 Jan-Dec;15(1):2180315.