






联合scRNA-seq、MeRIP-Eseq、RNA-seq和Ribo-Eseq,7张图为你揭示M6A阅读器YTHDF1在抗结直肠癌免疫中的作用

N6-α-甲基腺苷(m6 A)是最丰富的RNA修饰之一,每个mRNA分子上估计有3-5个m6 A位点。M6A的甲基化修饰是甲基化转移酶,去甲基化,甲基化阅读蛋白共同参与。YTHDF1在m6A修饰中是作为甲基化阅读器,已经在许多研究中被证明YTHDF 1的上调与各种癌症类型中的不良预后相关。本研究中,作者首先发现了YTHDF1表达与IFN-γ的表达和CD8+T细胞浸润呈负相关,暗示了YTHDF1在结肠癌肿瘤微环境中的作用。通过在肿瘤模型上验证和scRNA-seq、MeRIP-Eseq、RNA-Eseq和Ribo-Eseq,证明了通过m6A-p65-CXCL 1/CXCR2轴抑制抗肿瘤免疫以促进结肠癌进展。该研究于2023年1月发表在《Gut》上,IF:24.5。
主要研究结果
1.YTHDF1与结肠癌预后不良相关以及单细胞测序揭示YTHDF1介导的免疫抑制
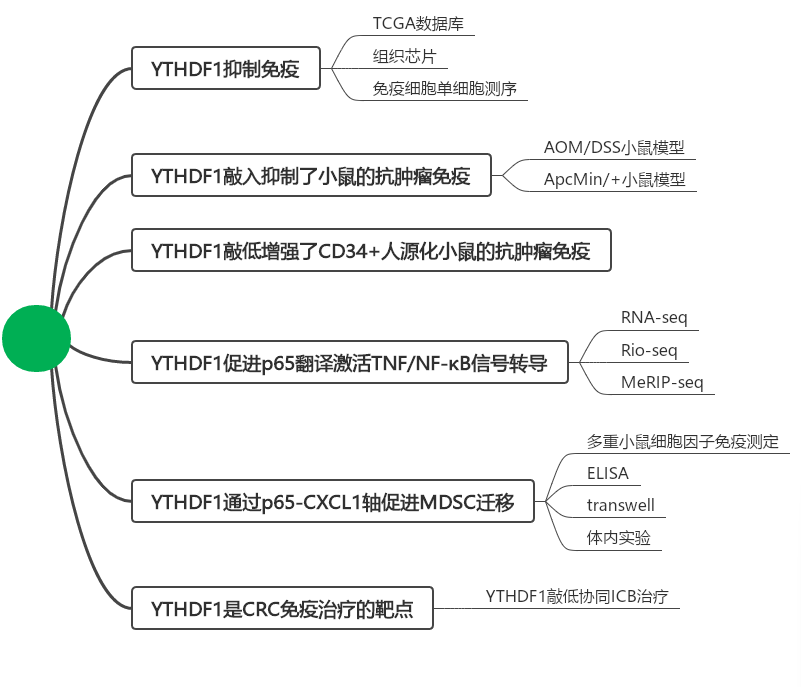
在80%以上的结肠癌(CRC)中,YTHDF1都表现为明显的升高,暗示YTHDF1在促进CRC中起作用。为了建立抗肿瘤免疫与m6 A调节子之间的联系,分析m6A调节子与IFN-γ应答基因和CD8+T细胞浸润之间的相关性,通过TCGA数据库以及组织芯片染色分析,发现YTHDF 1与IFN-γ应答途径和CD8+T细胞浸润呈显著负相关(图A、B和C)。为了研究YTHDF1在调节抗肿瘤免疫中的作用,研究者使用CRISPR-cas9系统敲除MC38鼠MSI-casH CRC细胞中的YTHDF1(YTHDF1-cas9 KO),并将细胞注射到同基因C57BL6小鼠中(图1D)。与对照相比,通过敲除YTHDF1l降低了肿瘤体积和重量(图1E)。从肿瘤中分离CD45+免疫细胞并进行单细胞RNA-seq(scRNA-seq)。与NC组相比,具有YTHDF1-KO的肿瘤表现出粒细胞髓系-衍生的抑制细胞(G-MDSC,簇3)和嗜中性粒细胞的显著减少(图1F)。进一步将T细胞和NK细胞重新聚类为CD4+T、CD8+T、NKT和NK细胞亚群,与对照相比,它们在YTHDF11-KO肿瘤中同时增加(图1F)。检查了MDSC的功能标志物Illb、Arg2、Cxcr2和Ccr2,发现这些基因主要富集在MDSC簇(簇3和簇4)中,特别是来自YTHDF1未敲除的肿瘤(图1G)。通过流式细胞术测定MC38同基因小鼠中肿瘤浸润免疫细胞的组成。我们证实YTHDF1敲除显著抑制肿瘤重量和体积(图1C),并且流式细胞术揭示YTHDF1敲除减少MDSC,但增加肿瘤中的CD8+T和CD4+T细胞(图1H,I)。在MDSC中,G-MDSC是主要的亚群,并且YTHDF1的敲除导致G-MDSC的显著减少(图1 I)。这些结果证明了YTHDF1在CRC中的免疫抑制功能。
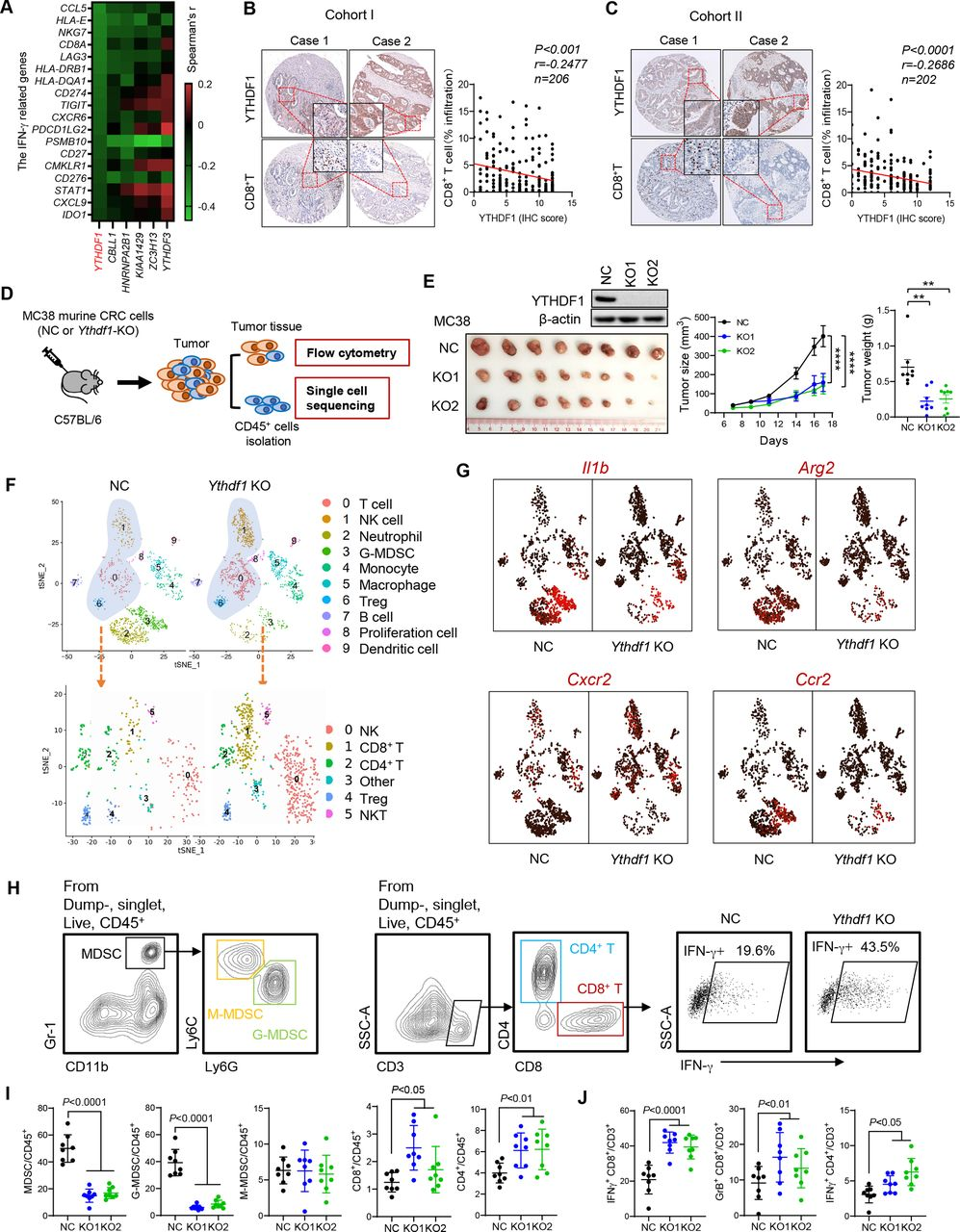
图1:YTHDF1与CRC患者中的免疫微环境相关
2.YTHDF1敲除减少了MDSCs细胞并增加了细胞毒性T的浸润
接下来在CT26(MSS-CRC小鼠模型)中用YTHDF1-KO进行实验以验证YTHDF1在调节抗肿瘤免疫中的作用。如所预期的,YTHDF1-KO导致CT 26同基因小鼠中肿瘤体积和重量减少(图2A和B),以及功能性T细胞减少和MDSC积累减少(图2C和D)。免疫荧光染色证实了在YTHDF1敲除后MC38和CT26同源肿瘤中MDSC(CD11b +Gr-b1+)的浸润减少(图2 E)。总的来说,CRC细胞中的YTHDF1耗竭减少MDSC并增加功能性T细胞浸润。这些发现与临床数据一致,表明YTHDF1与CD8+T细胞和IFN-γ相关特征呈反相关。YTHDF1-KO中减弱的肿瘤形成是否依赖于CD8+ T细胞抗肿瘤免疫。为了解决这一点,又在MC38同基因模型中用抗CD8抗体耗尽CD8+T细胞。与上面的假设一致,CD8+T细胞的消耗恢复了YTHDF1-KO肿瘤的生长(图2F和G),表明YTHDF1-KO的肿瘤抑制功能至少部分依赖于CD8+T细胞。这CT26同基因小鼠中得到证实,显示抗CD8抗体治疗挽救了YTHDF1-KO组中停滞的肿瘤生长(图2 H,I)。总之,YTHDF1敲除通过诱导CD8+T细胞依赖性抗肿瘤免疫抑制CRC生长。
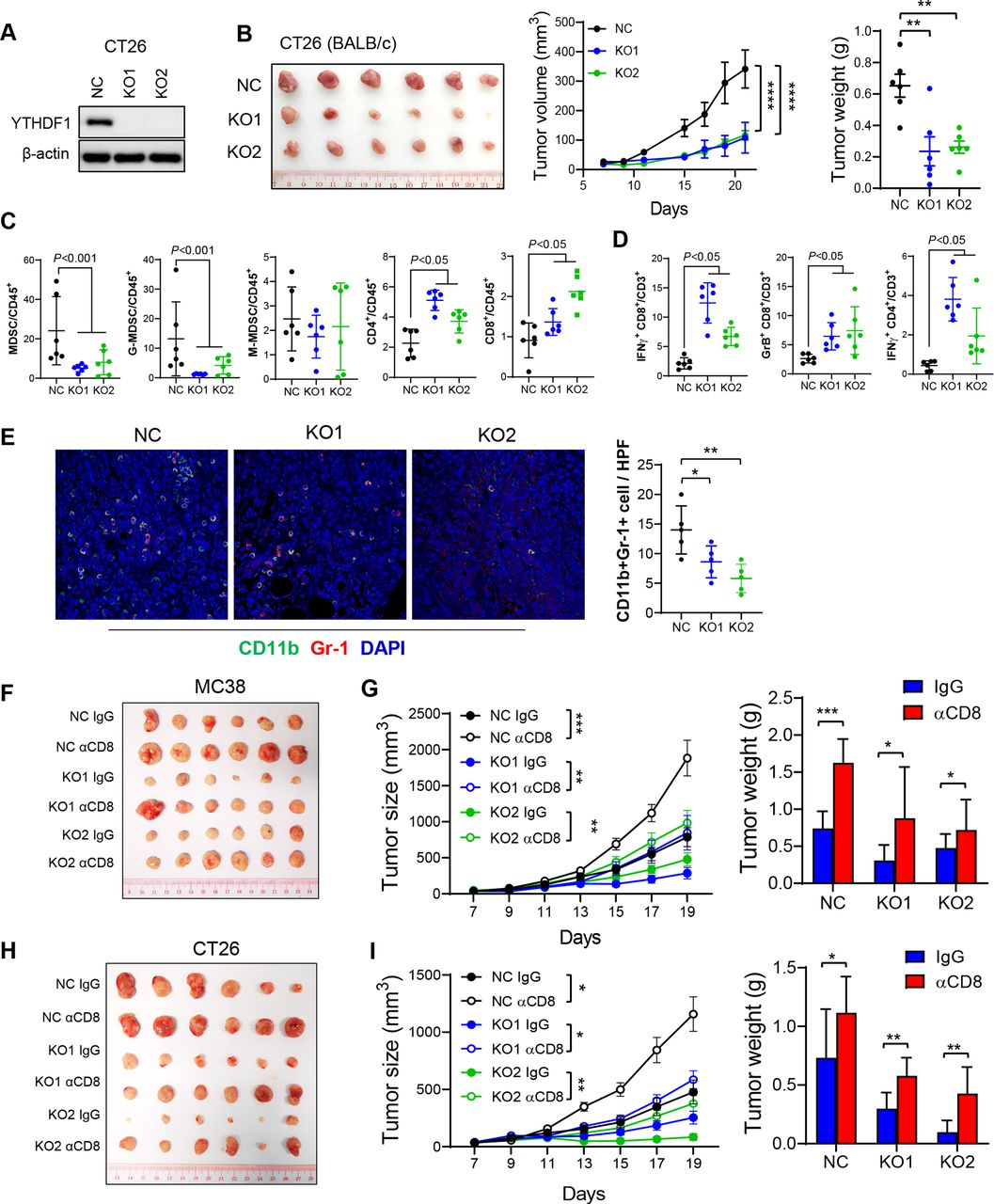
图2:YTHDF1敲除增加了MDSC和功能性T细胞来诱导抗肿瘤免疫
3.肠特异性YTHDF1基因敲入促进小鼠结直肠肿瘤发生并抑制抗肿瘤免疫
利用肠特异性YTHDF1敲入小鼠(YTHDF1loxp/loxp CDX2-cre),并通过AOM/DSS处理在这些小鼠中引发CRC(图3A)。发现YTHDF1的过表达导致AOM/DSS模型中结肠肿瘤数量和大小增加(图3C)。流式细胞术显示与野生型小鼠相比,YTHDF1敲入小鼠的结肠肿瘤中MDSC浸润增加以及NK、CD4+T和CD8+T细胞减少(图3D)。此外,还发现YTHDF1敲入降低了功能性T细胞的比例,如通过颗粒酶B、INF-γ和TNF-α表达所鉴定的(图3E)。为了验证ApcMin/+驱动的自发性CRC中的上述结果,又构建了ApcMin/+ YTHDF1loxp/loxpCDX2-cre小鼠模型,同样的,发现YTHDF1敲入的ApcMin/+小鼠中结肠肿瘤数量和大小增加(图3B)。肿瘤浸润性免疫细胞显示在具有ApcMin/+的肿瘤中NK、CD4+T和CD8+T细胞的浸润显著降低,同时敲入YTHDF1,并诱导MDSC(图3F)。此外,YTHDF1敲入减少ApcMin/+小鼠中的颗粒酶B+、INF-γ+或TNF-α+ T细胞(图3G)。总的来说,这些结果支持YTHDF 1促进促进自发性CRC的免疫抑制微环境。
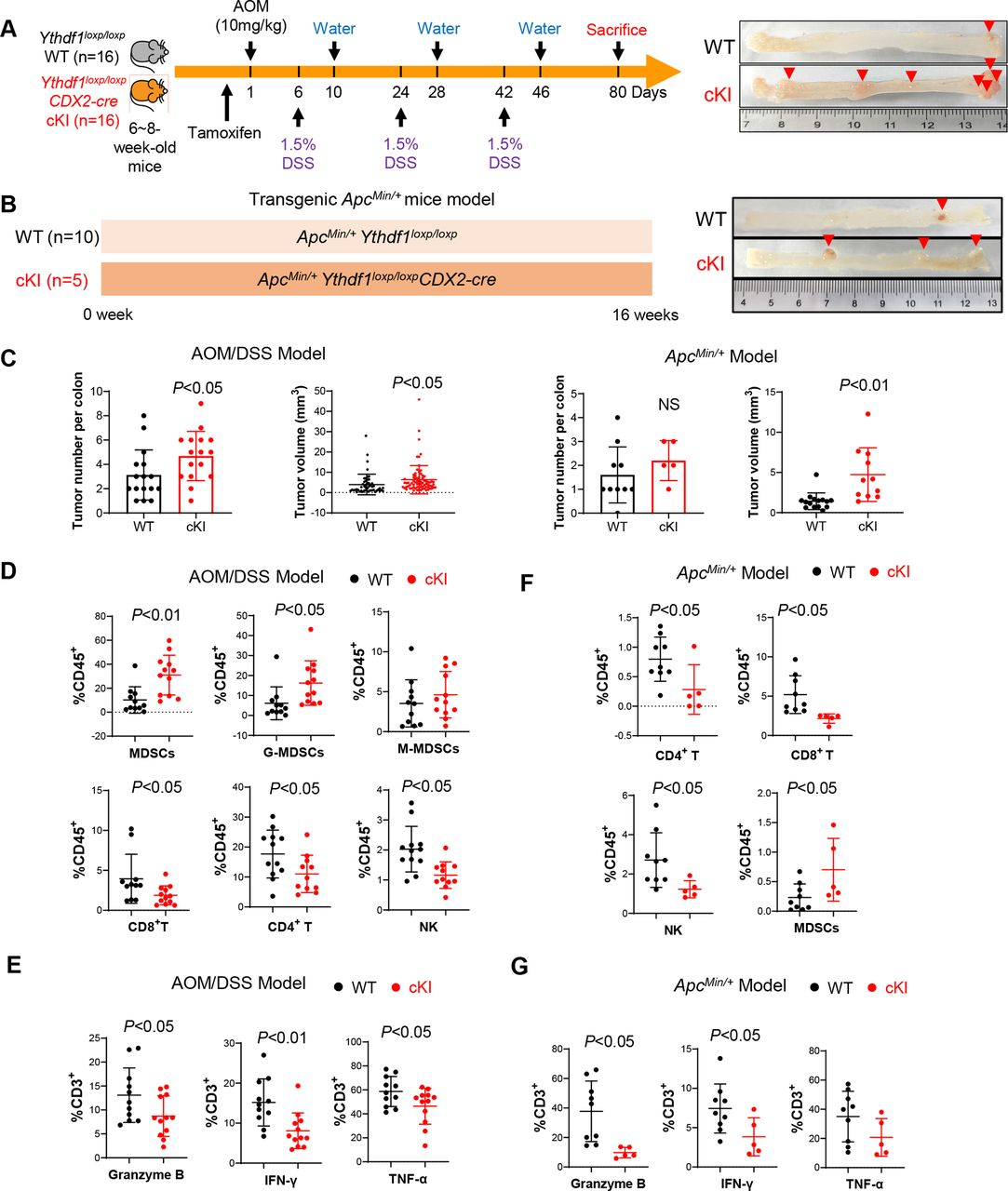
图3:YTHDF 1敲入促进小鼠结直肠肿瘤发生并抑制抗肿瘤免疫
4.通过VNP-siYTHDF 1靶向YTHDF1增强CD34+人源化小鼠的抗肿瘤免疫
构建CD34+人源化小鼠模型(图4A)。为了在体内靶向YTHDF1,开发了携带针对YTHDF 1的siRNA的VNPs 。在肿瘤达到50-100 mm3后,用VNP-siNC或-siYTHDF 1处理携带人CRC HCT116异种移植物的人源化NSG小鼠(图4 B)。与VNP-siNC相比,VNP-siYTHDF 1显著抑制肿瘤体积和重量(图4 B、C)。还进行流式细胞术以分析肿瘤微环境(图4D)。VNP-siYTHDF1减少MDSC浸润,但增加CD4+T细胞、CD8+T细胞和NK细胞积累(图4 E)。此外,在接受VNP-siYTHDF1的肿瘤中鉴定出更多的IFN-γ+、TNF-α+和颗粒酶B+ 、CD 8+T细胞(图4 E)。因此,使用VNP-siYTHDF1靶向YTHDF 1是增强人源化小鼠中抗肿瘤免疫力的有效的手段。
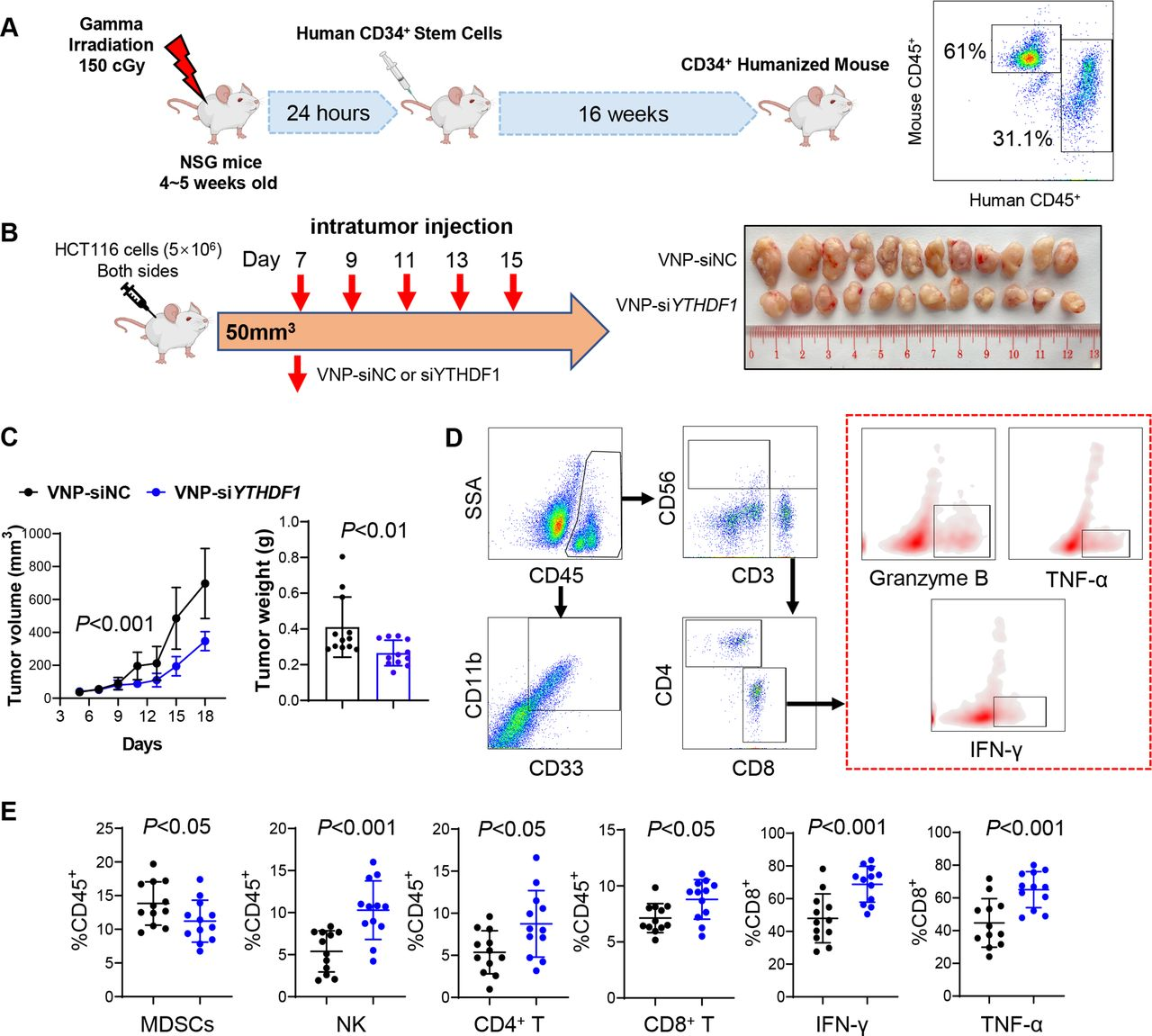
图4:VNP-siYTHDF1增强CD34+人源化小鼠中的抗肿瘤免疫
5.YTHDF 1促进p65翻译激活TNF/NF-κB信号转导
在敲除或未敲除YTHDF1的CRC细胞中进行了RNA-seq和Ribo-seq。通过RNA-seq分析,NC和YTHDF1-KO的MC38和CT26细胞之间差异表达的基因在TNF和NF-κB信号传导途径中富集。此外,Ribo-seq数据揭示YTHDF1的缺失与TNF信号传导的失活显著相关(图5A)。由于YTHDF1作为m6A阅读器起作用,接下来进行m6A免疫沉淀测序(MeRIP-seq)以精确定位m6A-seq修饰的转录物。通过筛选参与TNF信号传导的mRNA中的m6A峰,鉴定了两个接近p65 mRNA终止密码子的m6A位点(图5 B),并通过MeRIP-qPCR验证(图5C)。通过RIP-seq和使用抗YTHDF1抗体的RIP-qPCR鉴定了YTHDF1和p65 mRNA之间的直接相互作用(图5D)。敲除YTHDF1可减弱CT26和MC38细胞中p65蛋白表达,尤其是核p65表达,而不影响NF-κB通路的其他调节因子如IKKα和IκBα的表达(图5E)。在人CRC细胞中,显示野生型YTHDF 1的过表达,升高p65蛋白表达,相反地,YTHDF1敲低减弱人CRC细胞中的p65蛋白(图5 F)。使用抗YTHDF1抗体的RIP-α qPCR也证实了人CRC细胞中YTHDF1和p65 mRNA之间的直接相互作用(图5G)。验证YTHDF1和p65在体内的关联,将YTHDF1敲入小鼠中,发现结肠肿瘤中p65和磷酸化p65蛋白均增加(图5 H)。总之,上述的实验证明了YTHDF1促进p65蛋白的表达,以激活TNF和NF-κB信号传导。
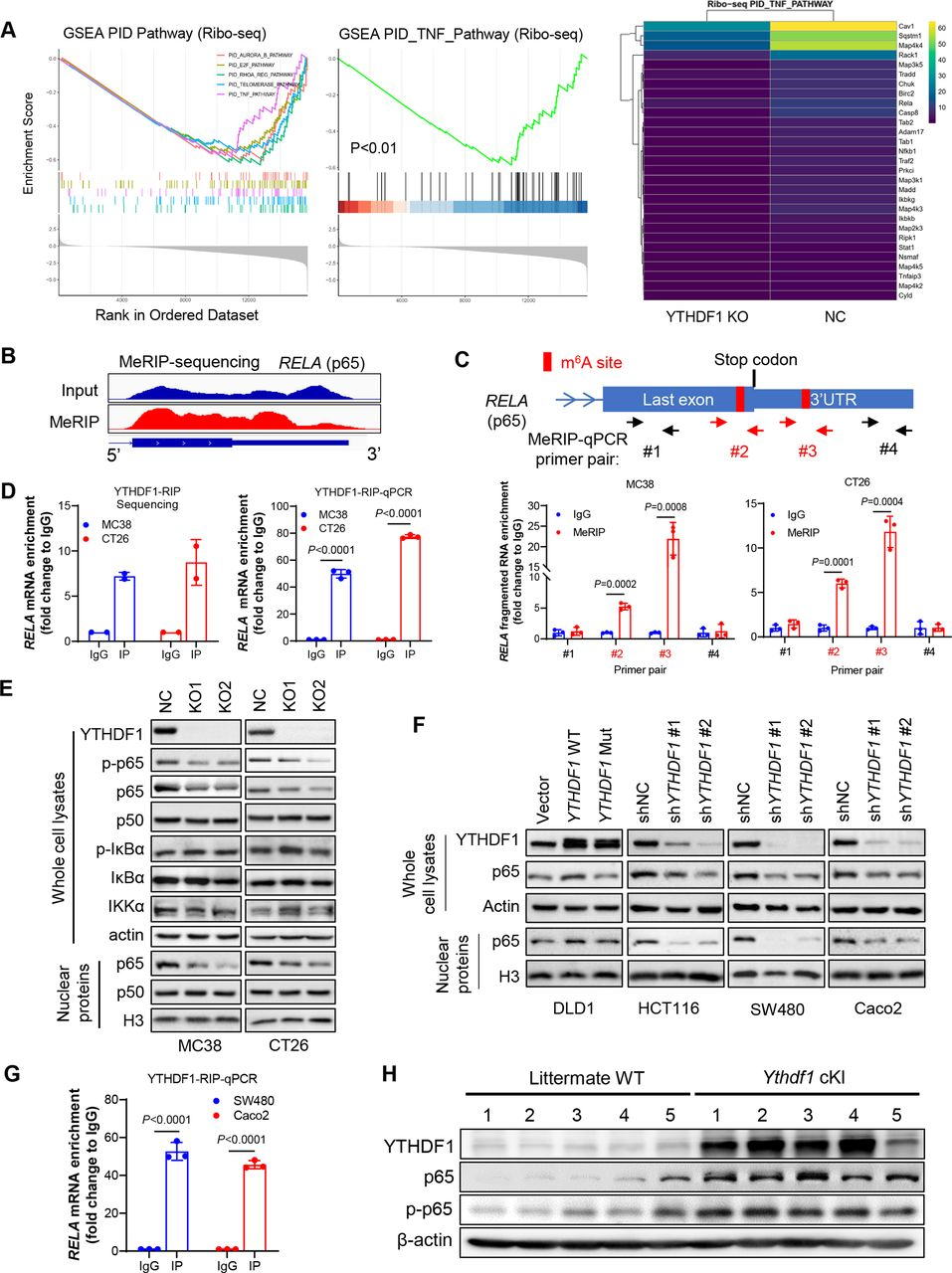
图5:YTHDF1通过促进p65mRNA翻译促进CRC中的TNF/NF-κ B信号传导
6.YTHDF1通过p65-CXCL 1轴促进MDSC迁移
进行细胞因子多重免疫测定,检测CRC细胞、肿瘤裂解物和来自携带同基因肿瘤的小鼠的血清的条件培养基中的23种不同的小鼠细胞因子。在这些细胞因子中,CXCL1在YTHDF1-KO组中一致地减少(图6A和B)。进行体外MDSC 迀移测定,发现来自野生型CRC细胞的条件培养基增强MDSC迀移(图6C)。通过CXCR2抑制剂阻断CXCL1/CXCR2相互作用,发现消除了对照和YTHDF1-KO培养物上清液在介导MDSC迀移方面的差异(图6C)。另外,YTHDF1敲除显著抑制鼠的CXCL1 mRNA水平(图6D)。接下来研究了YTHDF1是否影响MDSC功能。从MC38同基因肿瘤中分离CD11b+Gr- 1+ MDSC,然后与T细胞体外共培养(图6 E)。从对照肿瘤分离的MDSC抑制T细胞增殖。然而,与来自对照的MDSC相比,来自YTHDF1-KO肿瘤的MDSC对CD8+T细胞和CD4+T细胞的增殖表现出显著更低的抑制活性(图6 F和G),支持表达表达YTHDF1的CRC募集功能性MDSC。
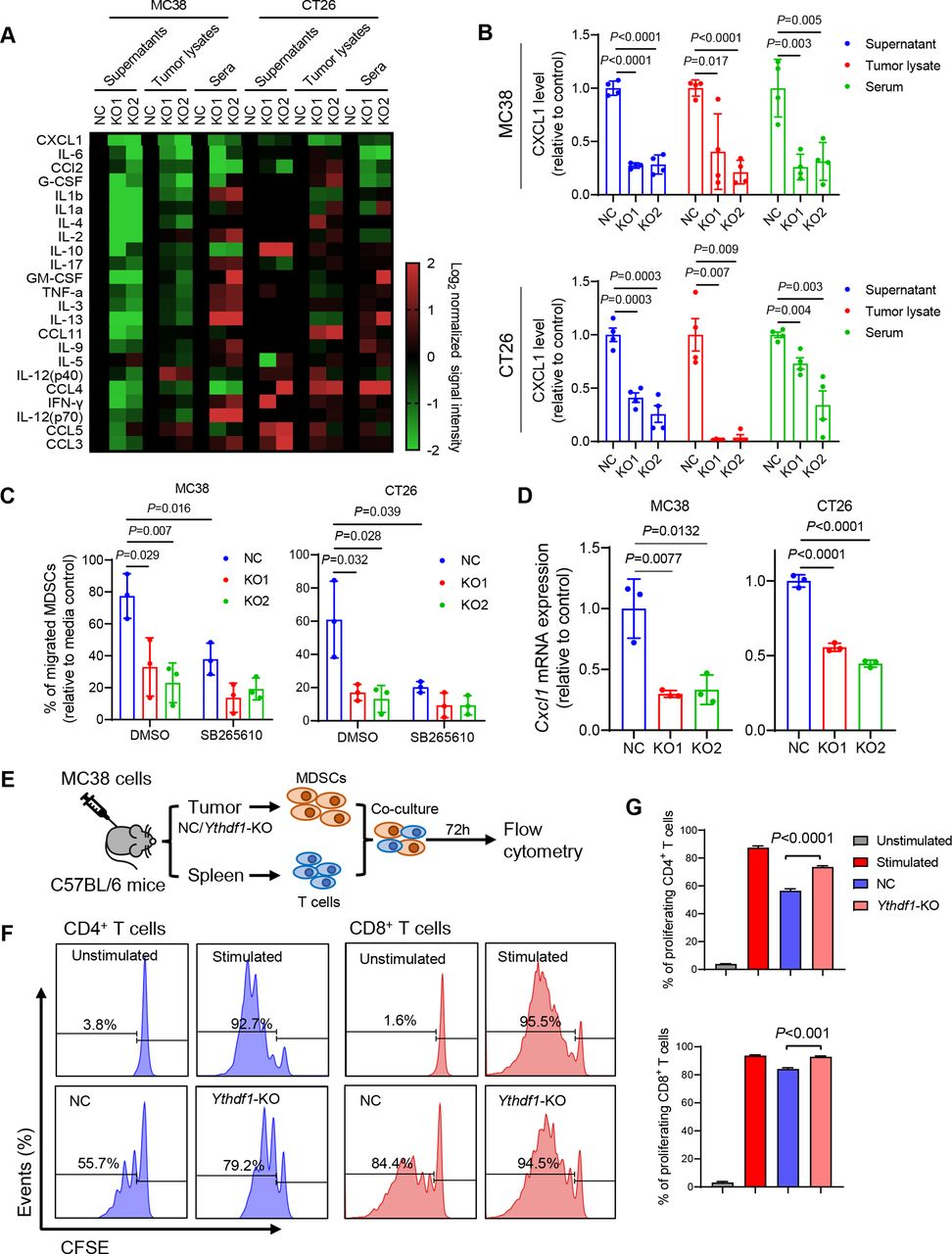
图6:YTHDF1通过调节CXCLl分泌介导MDSC的迁移
7.YTHDF1是结直肠癌免疫治疗的潜在治疗靶点
在小鼠中对YTHDF1的敲除,发现增强了抗PDl在MC38微卫星不稳定性高(MS1- H)同基因肿瘤中的功效,并延长了荷瘤小鼠的存活期(图7A)。当MC38同系肿瘤达到50~100 mm3时,用VNP-siYTHDF1(或VNP-siNC)和抗PD 1处理(或IgG)处理小鼠。与VNP-A siNC相比,VNP-siYTHDF1显著抑制MC38肿瘤生长(图7 B和C)。值得注意的是,VNP-siYTHDF1加抗PDl的组合对肿瘤生长发挥最强的抑制作用(图7 B和C)。基于同基因CT26(MSS-CRC)肿瘤模型,进一步解决了靶向YTHDF1是否可以克服MSS-CRC中的抗PD1抗性。因此,将具有YTHDF1敲除的CT26细胞注射到同基因小鼠中并用抗PDl处理。发现YTHDF1的敲除显著增强了CT26同基因肿瘤中的抗PDl治疗功效(图7 D和E)。流式细胞术分析进一步揭示,在CT26和MC38同基因模型中,YTHDF1沉默和抗PDl的组合显著增加肿瘤浸润功能性CD8+T细胞,包括IFN-γ+ CD8+T细胞和颗粒酶B+ CD8+T细胞(图7 F和G)。此外,组合治疗显著减少MDSC的积累,而诱导CD4+T细胞和CD8+T细胞(图7 G)。因此,这些结果表明YTHDF1的靶向不仅增强MSI-H CRC中的ICB治疗功效,而且还通过抑制MDSC的募集和改善CD8+T细胞的功能性来克服MSS-CRC中的ICB抗性。
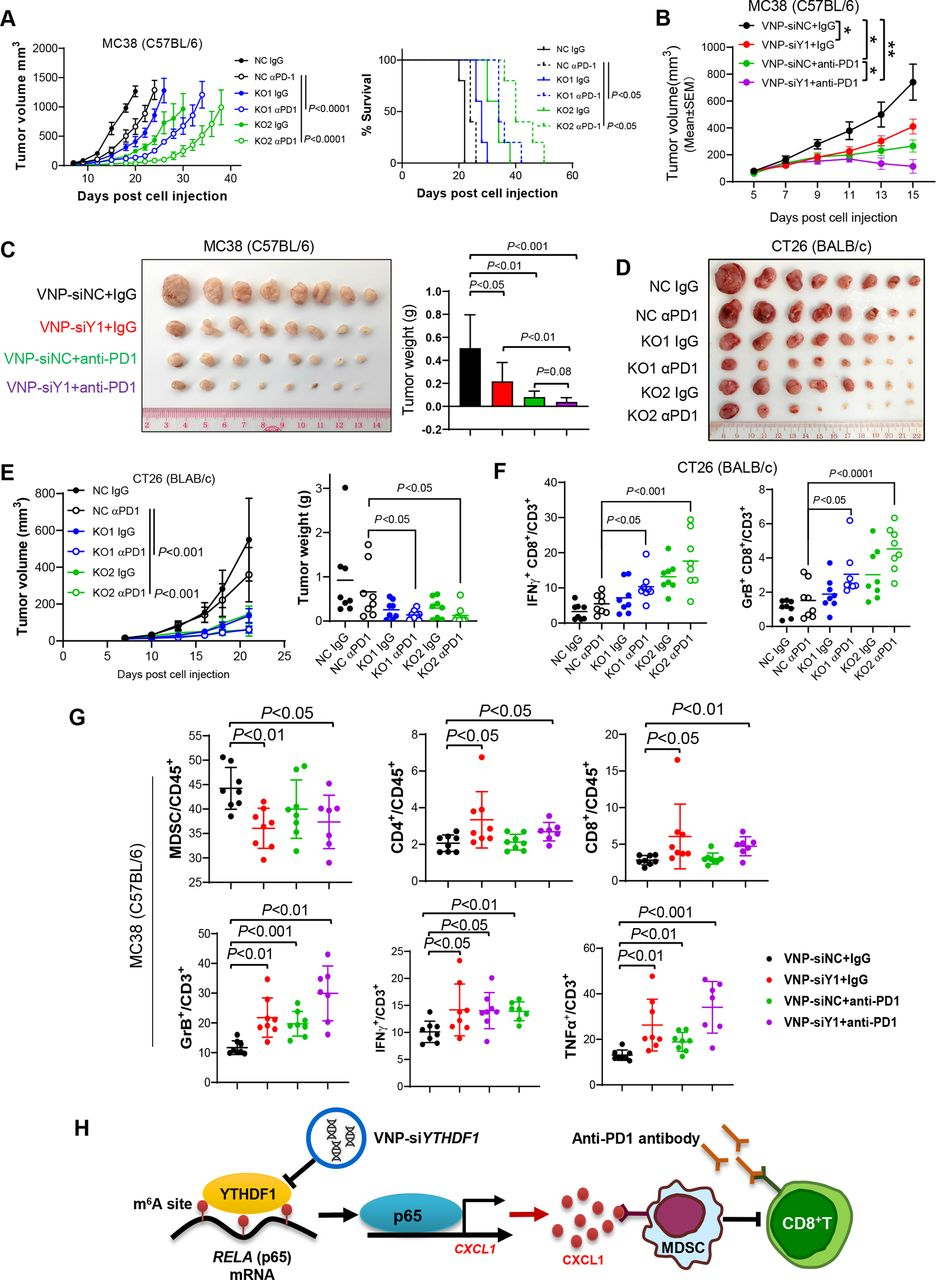
图7:YTHDF1增强了CRC中的抗PD 1阻断疗法
结论
综上所述,CRC中的YTHDF1表达通过激活m6A-p65-pCXCL1轴来募集免疫抑制性MDSC以抑制T细胞,从而促进CRC。靶向YTHDF1加抗PD 1治疗显示出针对CRC的抗肿瘤功效,证实YTHDF1是CRC的潜在治疗靶标。
实验方法
临床样本收集和收集细胞,TCGA数据分析,组织芯片,单细胞测序,AOM/DSS小鼠模型,Apcmin/+小鼠模型,人源化小鼠模型,RNA-seq,Rio-seq,MeRIP-seq,测序结果生信分析,多重小鼠细胞因子免疫测定,ELISA,transwell,western blot,RT-qPCR,. VNP-siNC和 VNP-siYTHDF1载体构建,细胞转染,流式细胞术,体内成瘤实验。
参考文献
Bao Y, Zhai J, Chen H, Wong CC, Liang C, Ding Y, Huang D, Gou H, Chen D, Pan Y, Kang W, To KF, Yu J. Targeting m6A reader YTHDF1 augments antitumour immunity and boosts anti-PD-1 efficacy in colorectal cancer. Gut. 2023 Aug;72(8):1497-1509. doi: 10.1136/gutjnl-2022-328845. Epub 2023 Jan 30. PMID: 36717220.