






铁死亡抑制剂可治疗大小鼠的糖尿病视网膜病
糖尿病视网膜病(DR)是导致全球失明的主要原因之一,及时预防和治疗非常重要。此前,作者发现一种神经退行性因子胶质细胞成熟因子-β(GMFB)在糖尿病早期就在玻璃体内上调,这可能在发病机制中起着重要作用。在这里,作者发现在高糖环境下,玻璃体内可分泌大量GMFB蛋白,它可将ATPase ATP6V1A 从溶酶体中转移出来,阻止其组装,并使视网膜色素上皮细胞(RPE)溶酶体碱化。ACSL4蛋白可被伴侣蛋白介导的自噬受体HSC70识别,并最终在溶酶体中被消化。自噬-溶酶体降解过程的异常会导致其积累,从而催化产生致命的过氧化脂质,最终诱发RPE细胞的铁死亡。GMFB抗体、溶酶体激活剂NKH477、CMA激活剂QX77和铁氧化酶抑制剂Liproxstatin-1都能有效预防早期DR,维持正常视功能,具有很强的临床应用价值。本研究拓宽了人们对自噬与铁蛋白沉积之间关系的认识,为治疗DR提供新的治疗靶点。本文于2022年7月发表在《Redox Biology》IF: 11.4期刊上。
技术路线

主要实验结果
1、玻璃体中高浓度的GMFB会损害视网膜和RPE细胞的功能
作者此前的研究结果表明,在糖尿病大鼠模型构建过程中,早在STZ注射后1天,玻璃体中GMFB就迅速增加(图1A)。为验证Müller细胞是否能在高葡萄糖刺激下分泌GMFB,作者用25 mM葡萄糖处理大鼠Müller细胞系,并在超滤离心后检测培养液中的GMFB含量。正如设想的那样,在高糖处理12小时后,培养基中的GMFB浓度明显增加(图1B)。为研究GMFB在体内的影响,将0.2 μg重组GMFB蛋白或等体积的PBS注入SD大鼠的玻璃体内,持续2/4 周。注射GMFB会明显破坏视网膜的生理功能并诱导神经变性(图1C-E),但不会激活神经胶质细胞(图1F-G)。鉴于已发现GMFB 可影响多种神经细胞的凋亡和氧化应激,作者还检测了注射GMFB后视网膜中的ROS水平,发现RPE细胞层是主要的作用部位(图1H)。
为在体外检测其影响,在GMFB处理ARPE19细胞后,检测ZO-1和RPE65 的表达模式以及TEER。如假设的一致,细胞相互作用或连接以及RPE65蛋白表达在暴露后有效减少(图1I-K)。总之,这些结果表明玻璃体内高浓度的GMFB会损害视网膜的生理功能,尤其是RPE细胞。
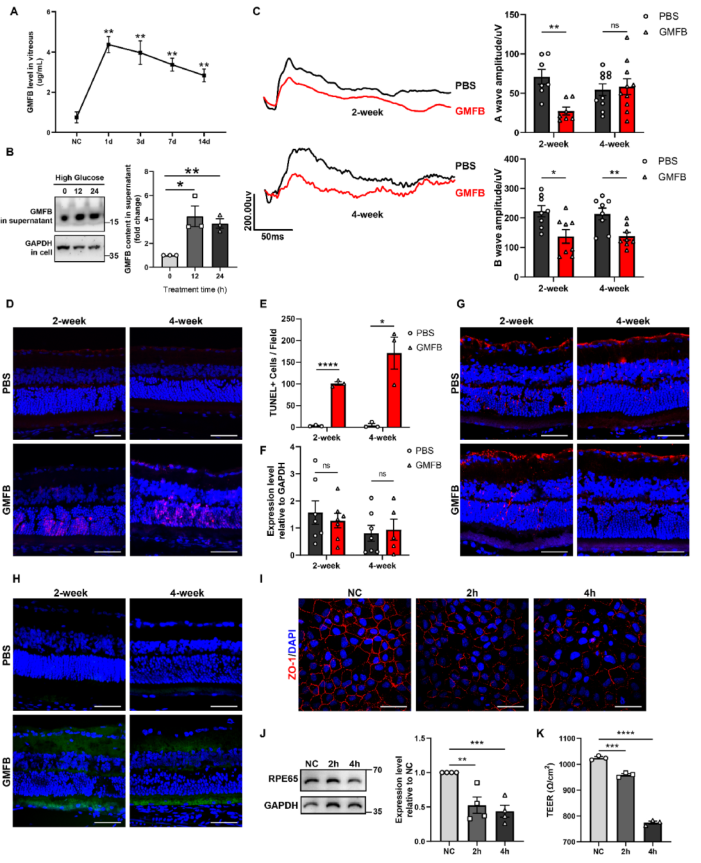
图1玻璃体中高浓度的GMFB会损害视网膜和RPE细胞的功能
2、细胞外GMFB诱导RPE细胞铁死亡
为确定与细胞外GMFB相关的基因组特征和机制,用GMFB处理ARPE19细胞4小时,并进行了基于RNA-seq表达的GSEA分析。意想不到的是,许多与铁死亡相关的通路在GMFB处理后的差异表达基因中得到了富集(图2A)。通过透射电子显微镜(TEM),观察到处理过的细胞中线粒体的形态特征明显变小,膜密度增加(图2B,红色箭头标记的线粒体),有报道称这种现象发生在铁死亡的细胞中。为进一步研究这一发现,检测细胞活力和两个公认标记物(ACSL4和GPX4)的蛋白表达水平,它们在GMFB处理后都发生显著变化(图2C-D)。
铁死亡的核心代谢机制是脂质过氧化和铁平衡失调。细胞外GMFB增加脂质过氧化、诱导GSH缺乏和线粒体活性降低(图2E-G)。与此同时,在注射GMFB的视网膜中也发现有毒的脂质过氧化产物4-HNE的积累(图2H)。为进一步研究GMFB是否通过脂质过氧化诱导的铁死亡影响细胞功能,用抑制剂铁死亡liproxstatin-1 (LX-1)预处理ARPE19细胞。LX-1阻止GMFB诱导的脂质过氧化、GSH缺乏和细胞死亡(图2C和4G),并对细胞连接有显著的保护作用(图2I-J)。这些结果表明,细胞外GMFB可诱导RPE细胞发生铁死亡反应,主要表现为脂质过氧化,并最终损害细胞的生理功能。
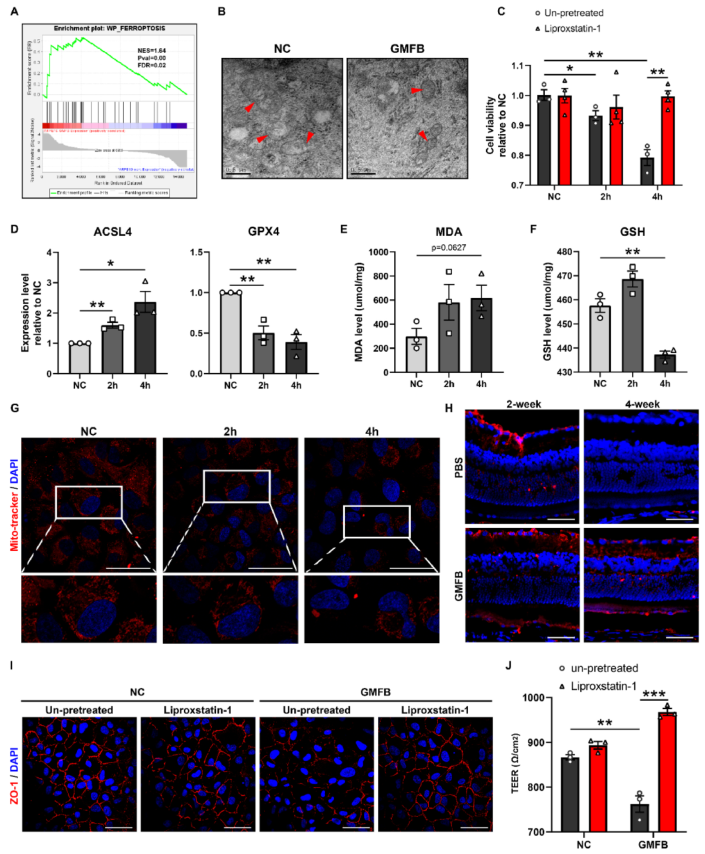
图2细胞外GMFB诱导RPE细胞铁死亡
3、细胞外GMFB诱导RPE细胞溶酶体功能障碍和自噬阻断
除线粒体受损外,还意外地在经GMFB处理的ARPE19细胞中用TEM观察到大量自噬空泡(图3A)。自噬降解脱落的感光细胞外节是RPE细胞最重要的功能之一。自噬体数量和P62蛋白表达的增加验证了GMFB对自噬通量的影响(图3B-C)。与体外实验结果一致,注射GMFB后,视网膜尤其是RPE层积累了大量自噬相关蛋白(图3D-F)。
鉴于P62的降解标志着自噬的正常功能,GMFB可能会影响ARPE19细胞自噬的后期阶段,包括自噬体-溶酶体融合和溶酶体功能。免疫荧光共定位结果未显示自噬体与溶酶体融合过程中的异常现象。然而,溶酶体追踪红染色结果表明,GMFB处理会破坏溶酶体的酸化和活性(图3G)。维持高酸性pH值对溶酶体的正常消化功能至关重要,而高酸性pH值主要由液泡H+-ATPase(V-ATPase)复合物产生。令人惊讶的是,经GMFB处理后,膜上亚基ATP6V1A的表达比例明显下降(图3H)。此外,ATPase激活 NKH47 增加了溶酶体的酸度,从而抑制了P62 的积累(图3G和4F)。这些数据共同表明,细胞外GMFB通过损伤RPE细胞溶酶体功能影响自噬通量。
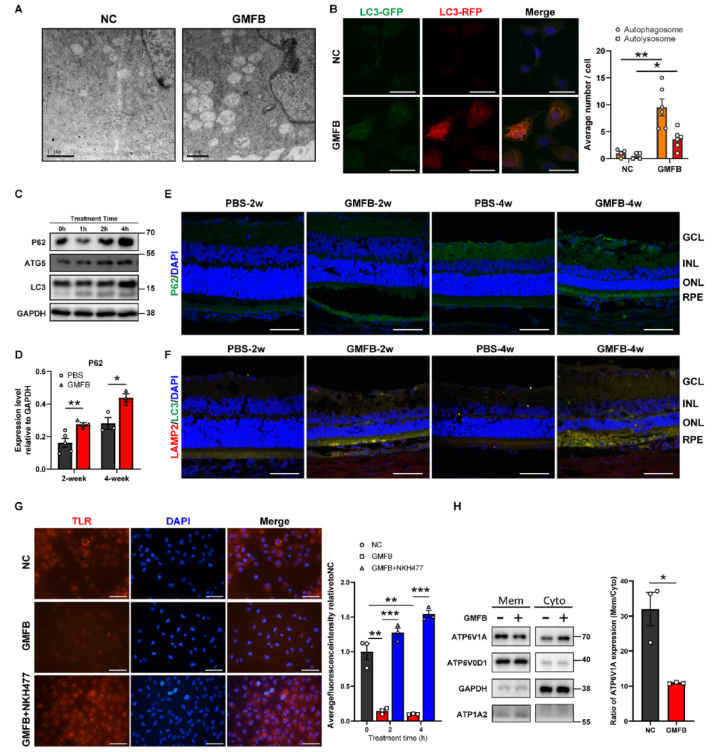
图3细胞外GMFB诱导RPE细胞溶酶体功能障碍和自噬阻断
4、GMFB诱导的溶酶体功能障碍通过ACSL4积累导致铁死亡
为确定溶酶体失效是否能诱导RPE细胞的铁死亡反应,直接用ATPase抑制剂BafA1处理细胞,它能特异性抑制溶酶体活性并诱导P62的积累(图4A)。如图4B-E所示,BafA1处理会显著降低细胞活力和GSH浓度,以时间依赖的方式增加ACSL4表达和MDA水平,并破坏RPE细胞的线粒体,所有这些都是铁死亡反应的特征。相反,ATPase激活剂NKH477增加溶酶体的酸度和活性,保护自噬通量,从而抑制P62和ACSL4的积累(图4F)。与假设类似,NKH477还能阻止GMFB诱导的细胞死亡、MDA生成和GSH缺乏,防止活性线粒体和细胞紧密连接的减少(图4G-K)。这些结果标志着溶酶体平衡在抵抗铁死亡反应中的重要性,并证明细胞外GMFB通过阻碍自噬-溶酶体降解诱导RPE细胞铁死亡反应。
假设溶酶体功能障碍通过上调ACSL4诱导铁死亡反应。令人惊讶的是,转染ACSL4-siRNA的细胞比对照组具有明显更高的活性和更低的MDA水平,即使在正常状态下,这些细胞也能抵抗GMFB或BafA1诱导的铁死亡反应(图4L-O)。这些结果表明,GMFB诱导的溶酶体失效通过上调ACSL4导致ARPE19细胞的铁死亡。
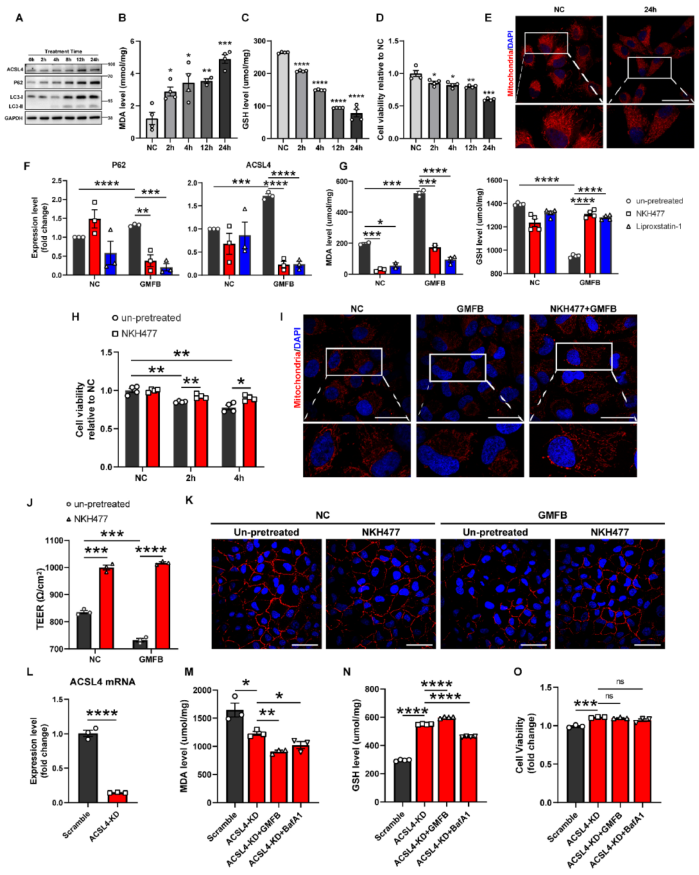
图4 GMFB诱导的溶酶体功能障碍通过ACSL4积累导致铁死亡
5、ACSL4是伴侣蛋白介导自噬(CMA)的底物
GMFB对ACSL4蛋白表达的影响不是通过激活转录实现的(图5A),这表明GMFB可能是通过损害溶酶体中的降解过程来上调ACSL4的。为验证这一点,用蛋白酶体或溶酶体抑制剂检测CHX处理后的蛋白表达(图5B)。然而,正如之前的假设,溶酶体抑制剂CQ也能抑制其降解(图5B)。这与之前研究的差异可能是由于细胞特异性或不同的疾病背景造成的。
自噬有三种形式:大自噬、微自噬和CMA,它们都在溶酶体中降解底物。在ARPE19细胞中,ACSL4没有与选择性大自噬所需的受体P62共定位(图5C)。然而,ACSL4与CMA受体LAMP2或HSC70之间存在明显的共定位,GMFB处理后这种共定位也增加了(图5D-E)。为研究ACSL4是否能被CMA降解,用不含FBS的培养液或含有QX77的完全培养液处理ARPE19细胞,QX77能通过增加LAMP2的表达激活CMA。与假设类似,这两种方法都促进了ACSL4的降解(图5F)。构建重组质粒ACSL4-Flag和HA-HSC70,通过免疫沉淀直接检测两者的结合。在ARPE19细胞的Flag-IP裂解中发现了HA-tag的表达,HSC70的加入也促进了ACSL4蛋白的降解(图5G-H)。在HEK293T细胞中也观察到了同样的结果(图5I)。
CMA只能降解与HSC70结合的、带有KFERQ样基序的可溶性蛋白质,而在ACSL4蛋白序列中可能有六个KFERQ样基序(图5J)。为了从功能上确定最重要的识别和降解基序,作者生成了在假定的CMA基团上有2-aa突变的ACSL4突变体:302QCERI306突变成302AAERI306,351QSSKI355突变成351AASKI355,407KLEQI411突变成407KLEAA411,566QIIDR570突变成566AAIDR570,574LVKLQ578突变成574LVKAA578,629QKGVE633突变成629AAGVE633。具有302QCERI306,566QIIDR570,574LVKLQ578的突变基序的ACSL4对HCC诱导的降解的抵抗性增强(图5K)。综上所述,ACSL4是伴侣蛋白介导的自噬的底物,可被受体HSC70识别并在溶酶体中降解。

图5 ACSL4是伴侣蛋白介导自噬(CMA)的底物
6、CMA激活剂QX77或铁死亡抑制剂LX-1可挽救GMFB诱导的视网膜功能障碍
为证实CMA和铁突变在GMFB诱导的视网膜病变中的作用,将GMFB与 CMA激活剂QX77和铁死亡抑制剂LX-1一起注入玻璃体内。这两种药物都能减少神经变性,并对视网膜的生理功能有显著的保护作用(图6A-B)。治疗后视网膜中4HNE的浓度和ACSL4蛋白的表达也有所下降(图6C-D)。意想不到的是,在注射GMFB组中,GPX4 的水平也有显著增加,而GPX4是一种抗氧化酶,在将有毒的脂质氢过氧化物转化为无毒的醇类方面起着关键作用。这可能是因为GPX4也可以通过CMA在溶酶体中降解,也可能是因为在复杂的视网膜结构中出现了代偿效应。
总之,本研究发现在高糖环境下,玻璃体内会分泌大量的GMFB蛋白,它能将ATPase ATP6V1A从溶酶体中转移出来,阻止ATPase ATP6V1A的组装,并使溶酶体碱化。ACSL4蛋白可被CMA受体HSC70识别,并最终在溶酶体中被消化。然而,降解过程的异常会导致其积累,从而促进脂质过氧化,最终诱发RPE细胞的铁死亡(图6E)。

图6 CMA激活剂QX77或铁死亡抑制剂LX-1可挽救GMFB诱导的视网膜功能障碍
为验证上述机制并探索治疗早期DR的新方法,作者建立了一个为期两周的糖尿病大鼠模型,并在检测到高血糖的第一天向玻璃体内注射GMFB抗体、溶酶体激活剂NKH477、CMA激活剂QX77或铁死亡抑制剂LX-1。令人惊讶的是,所有药物都能显著恢复视网膜的生理功能,使A波和B波几乎增加到正常水平(图7A-B)。此外,在注射药物组中,RPE-Bruch's膜-绒毛膜复合体(RBCC)中ACSL4的表达以及视网膜中的MDA水平均有所下降(图7C-D)。注射药物后,有报道称在糖尿病视网膜中积累的毒性脂质过氧化物4HNE也减少(图7E)。
此外,还通过免疫荧光染色P62和LC3来测量RPE层的自噬通量。包括LX-1在内的所有药物都显著减少自噬体的数量,这表明LX-1可能通过类似的途径抑制铁死亡(图7F)。GMFB抗体、NKH477、QX77和LX-1都能有效预防早期糖尿病视网膜病变,保护视网膜的正常功能。

图7 GMFB抗体、QX77、NKH477和LX-1对DR有保护作用
实验方法
糖尿病小鼠建模和药物处理,ELISA,免疫荧光,TUNEL,ERG检测视网膜功能,细胞培养,透射电镜,上皮电阻(TEER)测量,RNA测序,CCK8,脂质过氧化MDA实验,GSH实验,线粒体膜电位检测,溶酶体酸度检测,WB,mRFP-GFP-LC3细胞构建,siRNA基因敲除,质粒转染和位点突变,RT-qPCR
参考文献
Liu C, Sun W, Zhu T, Shi S, Zhang J, Wang J, Gao F, Ou Q, Jin C, Li J, Xu JY, Zhang J, Tian H, Xu GT, Lu L. Glia maturation factor-β induces ferroptosis by impairing chaperone-mediated autophagic degradation of ACSL4 in early diabetic retinopathy. Redox Biol. 2022 Jun;52:102292. doi: 10.1016/j.redox.2022.102292. Epub 2022 Mar 18. PMID: 35325805; PMCID: PMC8942824.